
基于点击化学的聚己内酯接枝共聚研究
2015-11-18 08:24:02李明明潘鹏举单国荣包永忠
化学反应工程与工艺 2015年3期
李明明,潘鹏举,单国荣,包永忠
化学工程联合国家重点实验室,浙江大学化学工程与生物工程学院,浙江 杭州 310027
聚己内酯(PCL)是一种典型的生物可降解高分子,降解后的产物为CO2和H2O,对环境和人体无危害。PCL 具有优良的生物相容性和力学性能,已被广泛用于药物缓释和组织修复等领域[1]。但PCL结晶性较强,亲水性较差,在体内降解速度较慢,从而限制了其应用领域。文献上对PCL 末端改性和嵌段共聚的研究报道较多,但嵌段共聚物结构与性能的均一性较差。近来,研究人员尝试在PCL侧链引入活性官能团以制备性能更加优异的接枝共聚物[2-4]。
本工作利用卤素官能化己内酯单体(CL)与未改性CL 的开环聚合(ROP),制备了侧链卤素官能化的PCL,然后将卤素转化为叠氮基团,再利用点击化学方法,将聚乙二醇(PEG)接枝至PCL主链,得到了PCL-g-PEG,该方法可有效调控共聚物的接枝密度和接枝链长度,同时研究了接枝共聚物在水溶液中的胶束化自组装,所制备胶束材料在药物缓释等领域具有潜在应用空间。
1 实验部分
1.1 主要原料
CL(>99%,TCI)、月桂醇(>99%,百灵威)、辛酸亚锡(95%,Aldrich-Sigma)、聚乙二醇单甲醚(mPEG,分子量1 000 和1 900,Alfa Aesar)、4-戊炔酸(95%,百灵威)、二环己基碳化二亚胺(DCC,99%,阿拉丁)、4-二甲基氨基吡啶(DMAP,99%,百灵威)、抗坏血酸钠(99%,阿拉丁)等均为分析纯。α-氯代CL(αClεCL)由Jerome 等所报道的方法制得[3]。CL 经氢化钙干燥48 h,减压蒸馏提纯。甲苯利用氢化钙为干燥剂,加热回流48 h 除水。
1.2 聚合反应
1.2.1 氯官能化PCL 的合成
利用CL 和αClεCL 开环共聚制备了氯官能化的PCL,即P(CL-co-αClεCL)。开环聚合中以月桂醇为引发剂,辛酸亚锡为催化剂,无水甲苯为溶剂。将一定量的αClεCL、CL、月桂醇和辛酸亚锡加入至经充分干燥的烧瓶中,其中催化剂与引发剂的物质的量比为3:1,将反应瓶连接至双排管,抽充氩气三次后注射加入甲苯。升温至70 ℃开始聚合,反应48 h,然后将产物溶于氯仿,缓慢滴至无水乙醇中沉淀以除去未反应的单体,沉淀三次,最后将粗产物在40 ℃真空干燥箱中干燥至恒重。氯官能化的PCL 用P(CL-co-xαClεCL)表示,x为制备中αClεCL 单体的投料质量分数。
1.2.2 叠氮官能化PCL 的制备
采用亲核取代法[4]制备了叠氮官能化PCL,即P(CL-co-αN3εCL)。将一定量的P(CL-co-αClεCL)、NaN3和N,N二甲基甲酰胺(DMF)加入到100 mL 烧瓶中,室温搅拌24 h,然后加入氯仿,用去离子水洗涤多次,有机相经无水硫酸镁干燥,旋转蒸发除去溶剂,然后产物在45 ℃下真空干燥至恒重。
1.2.3 末端炔化PEG 的制备
向250 mL 烧瓶中加入适量的mPEG、4-戊炔酸和二氯甲烷,将烧瓶置于冰水浴中,滴加DCC 的二氯甲烷溶液,同时缓慢滴加DMAP 的二氯甲烷溶液,其中mPEG、戊炔酸、DCC、DMAP 的投料物质的量比为1:1.2:1:0.05。反应物在0 ℃反应1 h,室温反应24 h,过滤,滤液依次用0.5 mol/L 的NaOH 水溶液、去离子水洗涤至中性,有机相经无水硫酸镁干燥,旋转蒸发除去溶剂,最后在45 ℃条件下真空干燥至恒重。
1.2.4 PCL-g-PEG 的制备
将适量的P(CL-co-αN3εCL)、mPEG、抗坏血酸钠、CuSO4加入至经充分干燥、充满氩气的烧瓶中,然后注射加入经氮气鼓泡除氧的DMF,室温下反应24 h,其中PCL 中叠氮基、PEG 中炔基、CuSO4催化剂、抗坏血酸钠还原剂的物质的量比为1:1.2:0.4:0.8。反应后将反应液注入透析袋(截留分子量为3 500)中,在去离子水中充分透析,最后将透析液冷冻干燥得到产物。接枝共聚物用PCLy-g-PEGz表示,其中y和z分别代表P(CL-co-αClεCL)制备中αClεCL 的投料质量分数和PEG 接枝链的分子量。
1.3 测试与表征
利用400 M NMR(Bruker AVANCE II)分析了样品的氢谱核磁(1H-NMR),以氘代氯仿或氘代水为溶剂,化学位移用溶剂峰标定。
利用Nicolet 5700 红外光谱仪测试了聚合物的红外光谱(IR)谱图。
利用Waters 凝胶渗透色谱仪(GPC,PL-gel mix C 色谱柱)测试了聚合物的分子量,流动相为DMF,流动相流速1.0 mL/min,测试柱温60 ℃,采用聚甲基丙烯酸甲酯作为标样。
采用表面张力法测定共聚物的临界胶束浓度(CMC)。将样品配制成不同浓度(4×10-4~1.0 g/L)的水溶液,然后在20 ℃条件下用表面张力仪测试了溶液的表面张力。CMC 由表面张力与样品浓度关系图求得。
胶束的流体力学直径(Dh)采用动态光散射(DLS,Zetasizer 3000 HAS)测试,测试前胶束溶液(1.0 g/L)用0.45 µm 有机过滤膜过滤。
透射电子显微镜(TEM,JEM-1230)观察胶束的结构形貌。将样品溶液滴至铜网上,置于真空干燥箱中室温干燥48 h,测试电压为80 kV。
2 结果与讨论
2.1 叠氮官能化PCL 的制备
图1为PCL-g-PEG 的合成路线。
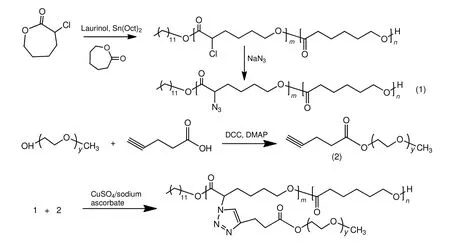
图1 点击化学法PCL-g-PEG 的制备Fig.1 Synthesis of PCL-g-PEG via click chemistry
首先利用开环聚合制备了P(CL-co-αClεCL),图2为P(CL-co-αClεCL)和P(CL-co-αN3εCL)的1H-NMR 与IR 谱图。
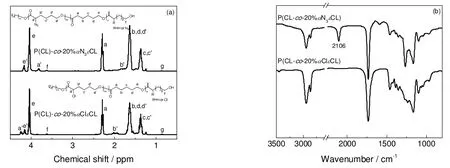
图2 P(CL-co-20%αClεCL)和P(CL-co-20%αN3εCL)的1H-NMR (a)和FT-IR (b)图谱Fig.2 1H-NMR (a) and FT-IR (b) spectra of P(CL-co-20%αClεCL) and P(CL-co-20%αN3εCL)
基于1H-NMR 的结果,计算了P(CL-co-αClεCL)的理论数均分子量(Mn,th)、αClεCL 单元的摩尔分数(nαClεCL)、质量分数(wαClεCL)和数均分子量(Mn,NMR),计算公式如下。
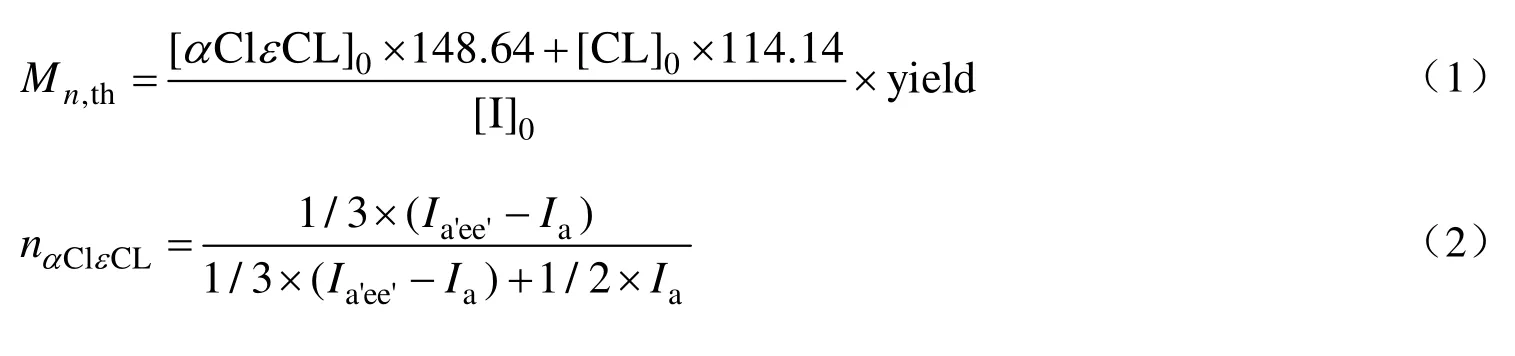

其中148.64 和114.14 分别为αClεCL 和CL 的摩尔质量,yield 为聚合物产率,I代表1H-NMR 谱图中对应峰的强度。式(2)中,在4.3 ppm (a′峰)和4.2 ppm (e′峰)处核磁共振峰分别代表αClεCL 中—CHCl—和—CH2COO—单元中氢的共振峰,与εCL 中—CH2COO—单元的氢的共振峰(a 峰)强度进行对比,可得到共聚物中αClεCL 单元所占的比例。
利用P(CL-co-αClεCL)与NaN3的亲核取代反应制备了P(CL-co-αN3εCL)。如图2(a)所示,在化学位移3.8 ppm 附近出现—CHN3—单元中氢的共振峰,同时P(CL-co-αClεCL)中化学位移4.3 ppm 附近处的—CHCl—单元中氢的核磁共振峰消失,说明氯完全被叠氮基团所取代。在图2(b)中,2 106 cm-1处出现了叠氮基团的伸缩振动峰,说明成功制备了P(CL-co-20%αN3εCL)。表1列出了共聚物的投料比、化学组成和分子量,由该表可知,wαClεCL与αClεCL 的投料比基本相同,GPC 所测数均分子量(Mn,GPC)与Mn,th、Mn,NMR非常接近,分子量分布指数较小,说明P(CL-co-αClεCL)的共聚组成和分子量可由开环聚合很好控制。
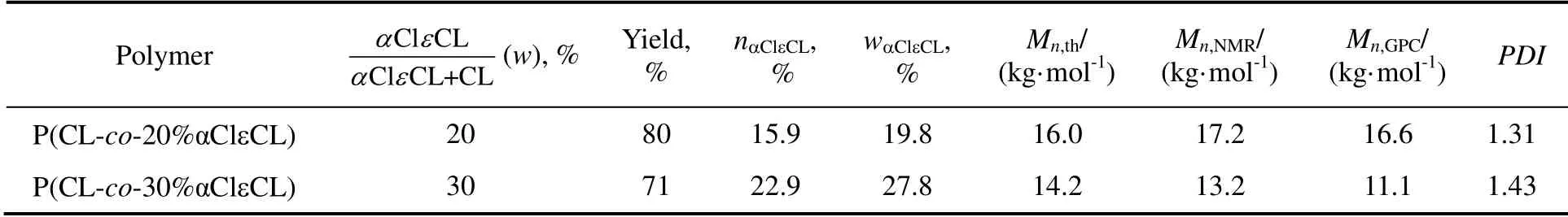
表1 P(CL-co-αClεCL)的合成投料比、共聚组成和分子量Table 1 Feed ratio, composition, and molecular weight of P(CL-co-αClεCL)
2.2 PCL-g-PEG 的制备
利用端炔基PEG 和P(CL-co-αN3εCL)的点击化学反应制备了PCL-g-PEG。如图3(a)所示,在化学位移7.6 ppm 附近可观察到唑环上次甲基的特征峰,在化学位移5.3 ppm 附近出现紧邻唑环的PCL 骨架上的次甲基的特征峰。如图3(b)所示,IR 图谱中2 106 cm-1附近处叠氮基团的伸缩振动峰消失,1 100 cm-1附近出现PEG 中C-O 键的伸缩振动峰。
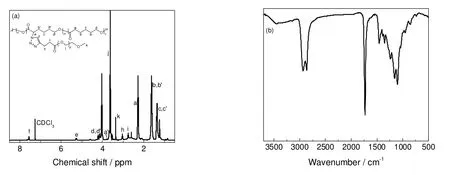
图3 PCL20-g-PEG1.0k 的1H-NMR (a)和FT-IR (b)图谱Fig.3 1H-NMR (a) and FT-IR (b) spectra of PCL20-g-PEG1.0k
如图4所示,随着接枝链长度增加,GPC 流出时间变短,共聚物分子量增大,NMR、IR 和GPC的结果均表明PEG 链已成功接枝至PCL 骨架。但是,如图3(a)所示,在化学位移3.8 ppm 处可观察到—CHN3—单元中氢的较弱的核磁共振峰,说明少量叠氮基未参加点击化学反应。
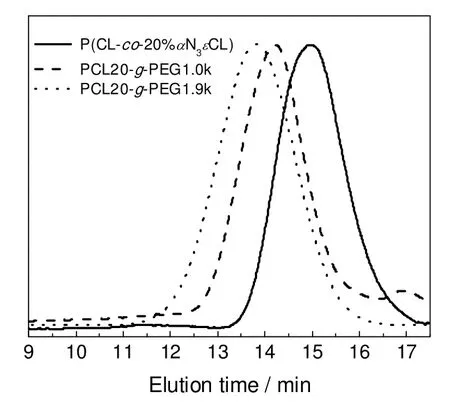
图4 含不同接枝链长度的PCL20-g-PEG 的GPC 结果Fig.4 GPC traces of PCL20-g-PEG graft copolymers with different graft lengths
表2列出了所制备PCL-g-PEG 的共聚组成、分子量和物理性质,其中DS表示点击化学中叠氮基团的反应百分率,Wgraft为共聚物中接枝链的质量分数,Mn,th表示接枝共聚物的理论数均分子量,其计算方法如公式(5)~(7)。式(5)中,Ie为点击反应后PCL 主链与唑环相连处CH2中氢的核磁共振峰强度(图3a),Ia′为未发生点击反应的PCL 主链与叠氮基团相连处CH2中氢的核磁共振峰强度(图2a)。式(6)中,Idd′代表PCL 骨架的—CH2OOC—单元中氢核磁共振峰的强度,Ij代表PEG 接枝链—OCH2CH2—单元中氢的核磁共振峰的强度,两者对比可得接枝共聚物的共聚组成。由表2可知,Mn,th和Mn,GPC较接近。由于共聚物中PEG 含量较大,其在室温下均可直接溶于水。
2.3 PCL-g-PEG 的胶束化自组装
PCL-g-PEG 具有两亲性,在水中可自组装形成胶束。通过比较PCL-g-PEG 在CDCl3和D2O 中的1H-NMR 谱图,确认了其胶束化[5]。如图5所示,在CDCl3中,PCL 和PEG 链段呈现清晰的共振特征峰,说明共聚物中所有链段均能很好地溶解。而在D2O 中,只能观察到亲水链段PEG 的特征峰,而疏水链段PCL 的特征峰较宽,并且非常弱,这表明在水溶液中PCL-g-PEG 形成以PCL 为核层、PEG为壳层的核-壳胶束。
采用表面张力法测定了PCL-g-PEG 的临界胶束浓度。当两亲性共聚物溶液浓度达到CMC 时,其表面张力与浓度的斜率发生突变,在CMC 之上随浓度的增大表面张力变化不明显[6]。如图6所示,CMC 由共聚物浓度与表面张力的关系曲线可以求得。共聚组成对PCL-g-PEG 的CMC 影响较大,当PEG 质量分数由47%增加至72%时,CMC由30.8 升高至65.6 mg/mL,这是因为共聚物中亲水性链段的含量升高所致。

图5 PCL20-g-PEG1.9k 在CDCl3 和D2O 中的1H-NMR 图谱Fig.5 1H-NMR spectra of PCL20-g-PEG1.9k in CDCl3 and D2O

图6 PCL20-g-PEG 水溶液浓度与表面张力的关系Fig.6 Plots of PCL20-g-PEG surface tension as a function of copolymer concentration
利用DLS 和TEM 研究了PCL-g-PEG 胶束的结构形貌。由表2可知,共聚物的Dh与其共聚组成有关。随着接枝链长度即PEG 分子量的增大,胶束粒径增大,这是由胶束的壳层较厚所致。以P(CL-co-20%αClεCL)为骨架的接枝共聚物为例,当PEG 分子量从1.0k 增加至1.9k 时,Dh从116.1 nm增大至136.1 nm。当PEG 链段的分子量一定时,接枝密度越小,胶束Dh越大,这是因为对于含有较多成核链段的共聚物,需形成较大核层的胶束才能包括所有疏水链段[7]。如图7所示,胶束粒径在100 nm 附近,但粒径分布较宽。如图8所示,胶束呈球形,粒径在100 nm 左右,但粒径分布较大,这与DLS 所测结果一致。
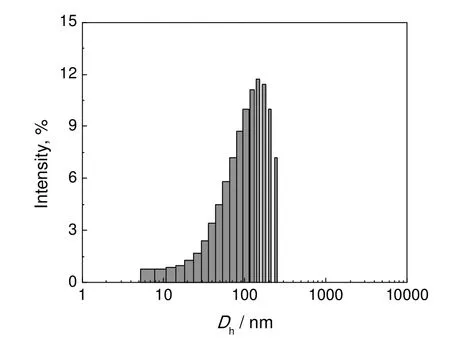
图7 PCL20-g-PEG1.9k 胶束粒径分布Fig.7 Particle size distribution of PCL20-g-PEG1.9k micelle

图8 PCL20-g-PEG1.9k 胶束的TEMFig.8 TEM micrograph of PCL20-g-PEG1.9k micelle
3 结 论
利用开环聚合制备了氯官能化的PCL,然后将氯转化为叠氮,进而采用点击化学法制备了PCL-g-PEG 接枝共聚物,并研究了其胶束化行为。开环聚合可良好控制氯官能化PCL 的分子量和氯官能基含量,1H-NMR 结果证明PCL-g-PEG 在水溶液中可自组装成球形胶束,随着PEG 接枝链质量分数的增大,PCL-g-PEG 的CMC值增大。PCL-g-PEG 共聚组成显著影响其粒径,在相同接枝密度时,Dh随着接枝链长的增大而增大。当接枝链长度一定时,接枝密度越大,Dh越小。
[1]Serrano M C, Chung, E J, Ameer G A.Advances and applications of biodegradable elastomers in regenerative medicine [J].Advanced Functional Materials, 2010, 20(2):192-208.
[2]Garg S M, Xiong X B, Lu C H, et al.Application of click chemistry in the preparation of poly(ethylene oxide)-block-poly(ε-caprolactone) with hydrolyzable cross-links in the micellar core [J].Macromolecules, 2011, 44(7):2058-2066.
[3]Lenoir S, Riva R, Lou X, et al.Ring-opening polymerization ofγ-chloro-α-caprolactone and chemical modification of poly(γ-chloro-α-caprolactone) by atom transfer radical processes [J].Macromolecules, 2004, 37(11):4055-4061.
[4]Riva R, Schmeits S, Jérôme C, et al.Combination of ring-opening polymerization and “click chemistry”:toward functionalization and grafting of poly(ε-caprolactone) [J].Macromolecules, 2007, 40(4):796–803.
[5]Guo S T, Wang W W, Deng L D, et al.Poly(ε-caprolactone)-graft-poly-[2-(dimethylamino) ethyl methacrylate]amphiphilic copolymers preparedviaa combination of ROP and ATRP:synthesis, characterization, and self-assembly behavior [J].Macromolecular Chemistry and Physics, 2010, 211(14):1572-1578.
[6]Kickelbick G, Bauer J, Huesing N, et al.Aggregation behavior of short-chain PDMS-b-PEO diblock copolymers in aqueous solutions [J].Langmuir, 2003, 19(24):10073-10076.
[7]Pan P J, Fujita M, Ooi W Y, et al.Thermoresponsive micellization and micellar stability of poly(N-isopropylacrylamide)-b-DNA diblock and miktoarm star polymers [J].Langmuir, 2012, 28(40):14347-14356.
猜你喜欢
合成技术及应用(2022年1期)2023-01-03 07:20:14
合成纤维工业(2022年2期)2022-05-06 12:03:08
商品与质量(2019年32期)2019-11-29 05:56:00
火工品(2018年1期)2018-05-03 02:27:56
中国资源综合利用(2017年3期)2018-01-22 02:45:40
天津农学院学报(2016年2期)2016-12-01 05:40:05
信息记录材料(2016年4期)2016-03-11 15:22:36
合成化学(2015年9期)2016-01-17 08:57:14
世界橡胶工业(2015年12期)2015-11-19 02:16:45
中国塑料(2015年8期)2015-10-14 01:10:53
