
磁性复合氧化物催化剂的制备及其催化木质素液化性能
2015-11-18 08:24:08谭方关包桂蓉李法社陈新怡
化学反应工程与工艺 2015年3期
谭方关,包桂蓉,李法社,陈新怡
省部共建复杂有色金属资源清洁利用国家重点实验室,昆明理工大学冶金与能源工程学院,云南 昆明 650093
由于化石能源过度开发使用导致全球气候变暖,可再生生物质能源日渐被重视。而可再生的木质纤维类生物质在自然界的大量存在确定了其作为替代能源的重要地位。木质素作为生物质的主要成分之一,由于结构复杂,不仅含有甲氧基、醇羟基和酚羟基等官能团,而且还有β-O-4 等醚键,α-O-4和4-O-5 等芳香醚键[1],所以其液化产物较为复杂。利用化学方法将木质素有效地催化液化成高品质液体燃料将是可再生能源一次重要的进展。
催化剂通常被用来在木质素液化过程中增加液体产物中高品位有机燃料的产率。现在的主流催化剂大多是贵金属催化剂、过渡金属盐、金属氧化物。其中贵金属催化剂具有较好的催化效果,钯催化剂与无催化剂相比在液化过程中能催化多产生超过10~20 倍的芳香醛[2]。Yan 等[3]在氢化处理的情况下,使用Ru/C、Pt/C、Rh/C、Pb/C 等催化剂在外加高压氢气下两步法分解白桦木材木质素转移加氢成烷烃(C8-C9,C14-C18),然而外加高压氢气不仅对设备有较高要求,而且工业运用也较为复杂。贵金属通常以固体颗粒状存在,便于回收和再利用,但贵金属的成本太高,Hasegawa 等[4-6]对金属氧化物催化剂做了深入研究,与过渡金属相比发现在木质素催化液化过程中金属氧化物催化剂表现出高的催化活性和稳定性。相比与贵金属,过渡金属盐的低成本更让人感兴趣,一般来说过渡金属盐通常具有水溶性,因此它们总是被作为均相催化剂。Bhargava 等[7]研究表明均相催化剂的催化活性由高到低的顺序为:Cu2+,Fe2+,Mn2+,Ce2+,Bi2+,Co2+,Zn2+,Mg2+,Ni2+。基于上述研究成果,为了使木质素能转化为液体燃料,Matson 等[8]利用Cu/Al2O3/MgO 催化剂实现了甲醇重整制氢和氢解纤维素同时反应的一锅法催化液化纤维素。Macala 等[9]利用Cu/Al2O3/MgO 在超临界甲醇中降解木屑取得了良好的效果。但是要将使木质素转化为高品质的液体燃料,通常还需要进行脱氧加氢反应,有研究表明MnO可作为脱氧加氢催化剂[10]。赵宁等[11]采用Cu/Mn/Ni/ZrO2催化剂合成低碳醇,其中锰是起增加组分在载体表面分散度的作用,并可保持催化剂的活性与稳定性,同时也有一定的促进产物碳链增长的能力。
在工业上,另一个较为困扰的问题是由于有效催化成分流失的均相催化剂的回收再利用[12],这些均相过渡金属盐会导致水排放的二次污染。在催化剂的使用过程中,使催化剂具备磁性能可以很好解决催化剂的回收再利用问题,而且能减少传统催化剂在回收利用过程中酸碱的使用。Zhang 等[13]为了能用磁场分离回收反应产物制备了Fe3O4@C-SO3H 催化剂,在140 ℃下反应12 h 催化分解纤维素,纤维素的转化率达到48.6%,葡萄糖的选择性达到了52.1%。因此,本研究采用共沉淀法制备Fe3O4@CuO/MnO/Al2O3作为催化剂,用于超临界甲醇中催化液化木质素,探讨焙烧温度对催化剂的物化性质、物相结构及催化反应性能的影响。
1 材料及方法
1.1 催化剂的制备
将 Cu(NO3)2·3H2O、Mn(NO3)2溶液(质量分数 50%)和 Al(NO3)3·9H2O 按物质的量比(Cu2+:Mn2+:Al3+为3:2:1)放入烧杯中,加入超纯水100 mL,室温下搅拌1 h 至完全溶解,得到的溶液标记为A;按计量配制的Na2CO3溶液标记为B;称取0.3 g 的纳米Fe3O4加入120 mL 超纯水,超声波振荡30 min,标记为C。将A 和B 分别装入滴定管中,以一定的速度同时滴入置于65 ℃恒温水槽的C 溶液烧杯中,同时搅拌使三种溶液充分混合,溶液pH 值恒定在9~10;滴加完毕后,混合溶液在65 ℃下继续搅拌1 h,然后室温下静置2 h。静置时,在烧杯底部放置磁铁,使混合溶液迅速沉淀分离,用超纯水多次清洗沉淀物直至清洗液显中性。清洗后的沉淀物在80 ℃下干燥12 h 得到层状氢氧化物催化剂前体(Fe3O4@CuMnAl-LDH),然后于N2保护的马弗炉中焙烧4 h 得到催化剂(Fe3O4@CuO/MnO/Al2O3)。
1.2 催化剂表征
催化剂的X 射线粉末衍射(XRD)在D/max-2200 型X 射线衍射仪上进行,Cu 靶,管电压和管电流分别为40 kV和40 mA,扫描速度为5 (o)/min。催化剂H2程序升温还原(H2-TPR)谱在Quantachrome公司ChemBET3000 型动态化学吸附仪上测定,催化剂用量10 mg,还原载气为5%H2/Ar,流速为40 mL/min,程序升温速率为10 ℃/min。热重(TG/DTG)分析采用德国NETZSCH 公司的STA 449 F3 Jupiter 同步热分析仪进行,流动N2氛围,流速40 mL/min,升温速率10 ℃/min。催化剂磁学性能采用美国Lake Shore 公司的7410 型振动样品磁强计(VSM),在室温下测量。气相色谱-质谱(GC-MS)采用TRACE DSQ 气相色谱-质谱联用仪进行分析,DB-5 柱子(30 mm×0.25 mm×0.25 μm);He 载气,流量1.0 mL/min;EI 离子源,离子源温度200 ℃;升温程序:起始35 ℃保持3 min,5 ℃/min 升温到60 ℃后保持2 min,然后10 ℃/min 升温到150 ℃,再20 ℃/min 升温到280 ℃。采用Wiley7n.l标准谱库计算机检索定性。液相产物中各组分百分含量由面积归一法计算得到。
2 结果与讨论
2.1 催化剂前体的表征结果
图1为Fe3O4@CuMnAl-LDH 的XRD 谱。在2θ为11.8,23.79,43.03 °时存在层状氢氧化物(LDH)的衍射峰,在2θ为31.15,35.45,62.56 °存在Fe3O4的衍射峰,符合典型的磁性水滑石特征峰特点[14]。

图1 Fe3O4@CuMnAl-LDH 的XRD 图谱Fig.1 XRD spectra of Fe3O4@CuMnAl-LDH

图2 Fe3O4@CuMnAl-LDH 的热重分析Fig.2 TG/DTG curve of Fe3O4@CuMnAl-LDH
图2是Fe3O4@CuMnAl-LDH 的热重分析结果。参考相关文献[15],170 ℃左右的快速失重峰归属于水滑石层间结构水的脱除;320 ℃左右的失重峰归属于大量水滑石层间结构内氢氧根离子和部分碳酸根离子的失去;620 ℃左右的失重峰归则归属为(Cu,Mn)AlxOy(CO3)z分解脱除CO2,故属于Fe3O4@CuMnAl-LDH 的层状氢氧化物的结构被破坏,故催化剂前体在此温度焙烧时会导致催化剂的结构发生不可逆转的变化。
图3是Fe3O4@CuMnAl-LDH 的SEM 图。从图中可以看出,黑色部分是纳米Fe3O4粒子,灰色部分是CuMnAl 水滑石,而且灰色部分比较均匀地覆盖在纳米Fe3O4粒子的表面[16],从而形成了大小均一、成分均匀的磁性催化剂前体,而且由无数个小层板重叠形成类似海绵状的层状结构[17]。

图3 Fe3O4@CuMnAl-LDH 的SEM 图谱Fig.3 TEM images of Fe3O4@CuMnAl-LDH
2.2 催化剂的表征结果
图4为不同焙烧温度下的催化剂的XRD 图谱。在33.2,35.5,49.3,53.8 °出现CuO 衍射峰,35.5 °时强度最大;在43.9,57.5,63.2 °出现较高角度的Fe3O4衍射峰;在21.5,30.2,44.7 °有CuAl2O4的衍射峰。焙烧温度低于450 ℃时,CuO 和CuAl2O4的衍射峰十分弥散;焙烧温度高于450 ℃时,随着焙烧温度的升高,CuO、Fe3O4的衍射峰不断锐化,但是CuAl2O4的衍射峰减弱。只有焙烧温度为450 ℃时,CuO、Fe3O4、CuAl2O4的衍射峰都比较明显,说明它们的结晶效果最好。另外,在所有样品上均未观测到MnO 的衍射峰,表明MnO 高度分散。
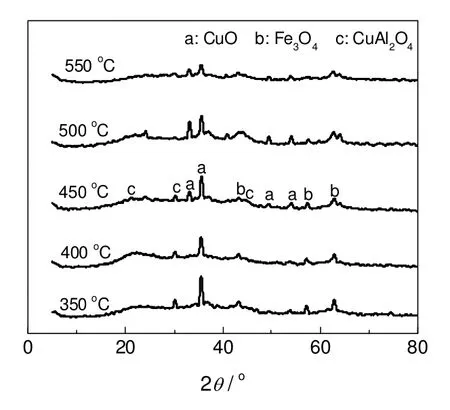
图4 不同焙烧温度制得催化剂的XRD 图谱Fig.4 XRD spectra of catalysts calcined under different temperatures
图5是不同焙烧温度制备催化剂的H2-TPR 谱。焙烧温度为400 ℃时,主还原峰出现在360 ℃处;焙烧温度大于400 ℃时主还原峰向低温偏移,均出现在300~320 ℃。这些还原峰均为单一对称峰,说明可还原的Cu 物种存在形式单一。结合图2的热重分析结果以及图4的XRD 分析结果,可将这些尖锐且峰形对称的还原峰归属为CuO 的贡献[18]。当焙烧温度大于450 ℃时,催化剂的还原峰峰高逐渐减小导致还原耗氢量减少。而且此后CuAl2O4尖晶石相的生成与水滑石结构分解析出CuO 同时发生,使得CuO 粒子被尖晶石相均匀隔离,且CuAl2O4尖晶石相是来源自Fe3O4@CuMnAl-LDH 的一次产物,而非析出的CuO与结构中的Al 氧化物形成的二次产物,因而对CuO 粒子主要起到物理分散作用。相比而言,当焙烧温度为450 ℃时,催化剂的还原峰温度较低,峰面积相对较大,此时催化剂的CuO 粒子被尖晶石相均匀隔离从而催化性能最好。此外从H2-TPR 图谱中还可以看出,在560~640 ℃出现一个强度较弱的副还原峰,应归属为Fe3O4的贡献。
表1列出了经过不同温度焙烧后的Fe3O4@CuO/MnO/Al2O3的比表面积。从表中可以看出,在450 ℃焙烧时,催化剂的比表面积最大,当焙烧温度高于450 ℃时,随着温度的升高,比表面积不断减小。结合XRD 的分析结果可知,随着焙烧温度的升高,颗粒不断聚集,催化剂的晶型不断增大,导致催化剂的比表面积不断降低。

图5 不同焙烧温度催化剂的H2-TPR 图谱Fig.5 H2-TPR spectra of catalysts calcined under different temperatures

表1 不同焙烧温度制得催化剂的比表面积Table 1 Specific surface area of catalyst calcined at different temperature
2.3 焙烧前后催化剂的磁学性能分析
根据上述结果,选择焙烧温度为450 ℃的催化剂,对催化剂前体及催化剂的磁学性能进行对比分析,两者均呈现出较大的磁滞回曲线,结果如图6所示。此外催化剂焙烧前的饱和磁化强度为19.251 emu/g,矫顽力101.29 G,催化剂焙烧后的饱和磁化强度为17.501 emu/g,矫顽力为97.399 G。这些数据表明制备的催化剂具有优良的磁学性能,并在较宽的磁场范围内有较强的磁响应能力。催化剂磁场分离提取实验发现,反应三次后的催化剂仍然能被磁铁分离出来。
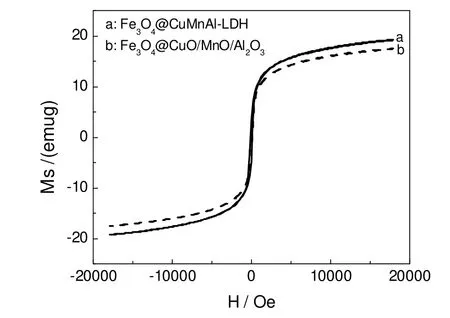
图6 焙烧前后催化剂的磁滞回曲线Fig.6 Magnetization curves of Fe3O4@CuMnAl-LDH and Fe3O4@CuO/MnO/Al2O3
2.4 不同焙烧温度催化剂的催化液化性能
采用不同焙烧温度的Fe3O4@CuO/MnO/Al2O3催化液化碱性木质素,以转化率为重要指标,优化催化剂的焙烧温度。在这组实验中,仅改变催化剂的焙烧温度,其他的液化条件相同,即木质素80 mg,催化剂30 mg,甲醇4 mL,在恒温340 ℃的锡池中反应120 min,结果如图7所示。从图7中可以看出,催化剂的焙烧温度对其催化效果影响显著。当焙烧温度在350~450 ℃时,木质素的转化率随焙烧温度的升高而提高,在450 ℃时达到最大值78.18%。焙烧温度大于450 ℃时,转化率不断下降。由此得出,在450 ℃的焙烧温度下,催化剂的催化液化效果最好,这与上述催化剂的H2-TPR、XRD 及比表面积的表征结果一致。

图7 不同焙烧温度制得催化剂催化木质素液化性能Fig.7 Liquefaction performance of lignin over catalysts calcined under different temperatures
2.5 液化产物分析
将木质素80 mg、450 ℃下焙烧的催化剂30 mg 以及甲醇4 mL 放入高压反应釜,在恒温340 ℃的锡池中反应120 min 后收集其液体产物,并采用GC-MS 进行检测,检测结果中的产物按族类分类分布如图8所示;其具体产物离子分布如图9所示,其中主要成分的含量为:醇类41.97%,酯类16.55%,烯烃1.43%,烷烃7.36%。醇类产物中主要为乙醇,其选择性高达19.6%,而烃类产物的选择性达到8.79%。与Matson 等[8]的研究相比,乙醇和烃类显著增多。

图8 超临界甲醇中Fe3O4@CuO/MnO/Al2O3 催化液化木质素产物分布Fig.8 Distribution of products by catalytic liquefaction of lignin in supercritical methanol

图9 超临界甲醇中Fe3O4@CuO/MnO/Al2O3催化木质素产物GC-MS 图谱Fig.9 GC-MS of products by catalytic liquefaction of lignin supercritical methanol
3 结 论
采用改进后的共沉淀法制备出大小均一,成分均匀的磁性催化剂前体Fe3O4@CuMnAl-LDH,CuMnAl 水滑石比较均匀地覆盖在纳米Fe3O4粒子的表面,形成了由无数个小层板重叠而构成的类似海绵状的层状结构。
催化剂前体在450 ℃下焙烧时分解,CuAl2O4尖晶石相的生成与水滑石结构分解析出CuO 同时发生,使得CuO 粒子被尖晶石相均匀隔离,易还原,因而催化剂的活性高,稳定性好,其催化性能最佳。此外该温度下焙烧得到的催化剂磁学性能优良而且稳定。
使用该催化剂在超临界甲醇中催化液化木质素,液化产物中醇类、烷烃类、烯烃类的相对含量分别为53.84%、1.83%与9.44%。反应产物中烃类的增加,说明催化剂的脱氧加氢作用明显。
[1]Strassberger Z, Alberts A H, Louwerse M J, et al.Catalytic cleavage of lignin beta-O-4 link mimics using copper on alumina and magnesia-alumina [J].Green Chem, 2013, 15(3):768-774.
[2]Sales F G, Maranhão L C A, Lima Filho N M, et al.Kinetic evaluation and modeling of lignin catalytic wet oxidation to selective production of aromatic aldehydes [J].Ind Eng Chem Res, 2006, 45(20):6627-6631.
[3]Yan N, Zhao C, Dyson P J, et al.Selective degradation of wood Lignin over noble-metal catalysts in a two-step process [J].Chem Sus Chem, 2008, 1(7):626-629.
[4]Hasegawa I, Inoue Y, Muranaka Y, et al.Selective production of organic acids and depolymerization of lignin by hydrothermal oxidation with diluted hydrogen peroxide [J].Energy &Fuels, 2011, 25:791-796.
[5]Deng H B, Lin L, Liu S J.Catalysis of Cu-doped Co-based perovskite-type oxide in wet oxidation of lignin to produce aromatic aldehydes [J].Energy &Fuels, 2010, 24:4797-4802.
[6]Deng H B, Lin L, Sun Y, et al.Activity and stability of perovskite-type oxide LaCoO3catalyst in lignin catalytic wet oxidation to aromatic aldehydes process [J].Energy &Fuels, 2008, 23(1):19-24.
[7]Bhargava S, Jani H, Tardio J, et al.Catalytic wet oxidation of ferulic acid (a model lignin compound) using heterogeneous copper catalysts [J].Ind Eng Chem Res, 2007, 46(25):8652-8656.
[8]Matson T D, Barta K, Iretskii A V, et al.One-pot catalytic conversion of cellulose and of woody biomass solids to liquid fuels [J].J Am Chem Soc, 2011, 133(35):14090-14097.
[9]Macala G S, Matson T D, Johnson C L, et al.Hydrogen Transfer from supercritical methanol over a solid base catalyst:a model for lignin depolymerization [J].Chem Sus Chem, 2009, 2(3):215-217.
[10]陈耀壮, 李 洁, 赵国强, 等.高效锰系脱氧剂的研制及性能测试[J].工业催化, 2013, 21(10):36-40.Chen Yaozhuang, Li Jie, Zhao Guoqiang, et al.Preparation and performance test of efficient Mn·deoxidizers [J].Industrial Catalysis,2013, 21(10):36-40.
[11]赵 宁, 陈小平, 魏 伟, 等.Cu/Mn/Ni/ZrO2合成低碳醇催化剂组分作用的初步研究 [J].燃料化学学报, 2001;29(S):146-9.Zhao Ning, Chen Xiaoping, Wei Wei, et al.Effect of component of Cu/Mn/Ni/ZrO2catalyst for higher alcohols synthesis [J].Journal of Fuel Chemistry and Technology, 2001, 29(S):146-149.
[12]Voitl T, von Rohr P R.Demonstration of a process for the conversion of kraft lignin into vanillin and methyl vanillate by acidic oxidation in aqueous methanol [J].Ind Eng Chem Res, 2009, 49(2):520-525.
[13]Zhang C B, Wang H Y, Liu F D, et al.Magnetic core-shell Fe3O4@C-SO3H nanoparticle catalyst for hydrolysis of cellulose [J].Cellulose, 2013, 20(1):127-134.
[14]Kannan S, Rives V, Knozinger H.High-temperature transformations of Cu-rich hydrotalcites [J].J Solid State Chem, 2004,177(1):319-331.
[15]汤 颖, 刘 晔, 路 勇, 等.CuZnAl 水滑石衍生催化剂上甲醇水蒸气重整制氢 I 催化剂焙烧温度的影响 [J].催化学报, 2006,27(10):857-862.Tang Yin, Liu Ye, Lu Yong, et al.Methanol steam reforming over CuZnAl catalysts derived from hydrotalcite precursor I effect of calcination temperature [J].Chinese Journal of Catalysis, 2006, 27(10):857-862.
[16]Tongamp W, Zhang Q, Saito F.Preparation of meixnerite (Mg-Al-OH) type layered double hydroxide by a mechanochemical route [J].J Mater Sci, 2007, 42(22):9210-5.
[17]Zhang F, Du N, Zhang R, et al.Mechanochemical synthesis of Fe3O4@(Mg-Al-OH LDH) magnetic composite [J].Powder Technol,2012, 228(3):250-253.
[18]Idem R O, Bakhshi N N.Production of hydrogen from methanol 2 experimental studies [J].Ind Eng Chem Res, 1994, 33(9):2056-2065.
猜你喜欢
天津医科大学学报(2021年1期)2021-12-05 11:11:05
上海包装(2019年8期)2019-11-11 12:16:14
天津造纸(2016年1期)2017-01-15 14:03:29
现代检验医学杂志(2016年5期)2016-08-20 03:17:08
中国塑料(2016年4期)2016-06-27 06:33:28
中国塑料(2016年11期)2016-04-16 05:25:56
中国造纸学报(2015年1期)2015-12-16 19:35:25
化学反应工程与工艺(2015年3期)2015-04-16 03:06:20
中国塑料(2014年4期)2014-10-17 03:00:45
技术与教育(2014年2期)2014-04-18 09:21:31
