
疏水改性聚磺酸甜菜碱的合成及其结构研究
2015-11-18 08:24:06马彦洁
化学反应工程与工艺 2015年3期
马彦洁,王 平,杜 淼,郑 强
高分子合成与功能构造教育部重点实验室,浙江大学高分子科学与工程学系,浙江 杭州 310027
疏水改性亲水性聚合物是指在亲水性聚合物主链上引入少量疏水基团的一类水溶性聚合物[1]。在水溶液中,疏水基团因疏水相互作用聚集在一起形成疏水微区,而疏水微区又可形成较大的缔合体,少量疏水基团的引入即可显著改变聚合物水溶液的流变行为。因此,通过疏水改性调控水溶性聚合物的结构进而控制其性能,引起了人们越来越多的关注[2,3]。据文献报道[4,5],该类聚合物具有明显的剪切增稠现象及优异的抗温抗盐性,因此在许多领域,尤其是石油开采的三次采油中广泛应用。
两性聚电解质是一类同时带有酸性和碱性基团的大分子。在适当条件下,这些基团可以解离,使分子链带正电荷或负电荷。一些蛋白质如明胶和牛血清蛋白,属两性聚电解质[6]。如果聚合物带有弱酸或弱碱基团,可通过改变溶液pH 值或外加小分子盐来调控其带电情况,改变聚两性电解质大分子的构象,进而影响其流变行为及使用性能。聚磺酸甜菜碱是甜菜碱衍生物,是一类重要的两性聚电解质,甜菜碱分子链中同时带有磺酸基和烷基取代铵基。疏水改性聚磺酸甜菜碱仍具有超亲水、抗蛋白吸附、环境友好等优异性能,使其在药物缓释、化妆品、器官移植、食品包装等领域具有巨大的潜在应用价值[7,8]。本研究将采用自由基胶束共聚方法[9],制备一种新型疏水改性聚磺酸甜菜碱,并探究聚合条件对共聚物结构及性质的影响。
1 实验部分
1.1 聚合反应
由亲水性单体[2-(甲基丙烯酰基氧基)乙基]二甲基-(3-磺酸丙基)氢氧化铵(SBMA)和疏水性单体甲基丙烯酸缩水甘油酯(GMA)通过自由基胶束共聚[9]合成疏水改性聚两性电解质P(SBMA-GMA)。将一定量SBMA 单体溶解在装有NaCl 水溶液的四口瓶中,称取一定量GMA 加到吐温(Tween)20的水溶液中,进行乳化后也加至四口瓶。氮气保护下搅拌0.5 h,然后加入一定量引发剂偶氮二异丁咪唑啉盐酸盐(VA-044),50 ℃下反应7 h。将粗产物用乙醇沉淀,以除去未反应的单体和乳化剂,然后用50 mL 的水将沉淀物重新溶解,上述过程重复三次。然后,用截留分子量为3 500 的透析袋进行透析、提纯一周,冷冻干燥获得最终产物。
1.2 表征方法
在红外光谱仪(Bruker Vector 22)上进行聚合物样品的红外(FT-IR)分析,采用溴化钾压片法。质量分数为10%的共聚物水溶液的表观粘度(ηa)在AR-G2 流变仪(美国TA 公司)上进行测试。用元素分析仪(Flash EA 1112)测试共聚物中各元素的含量。在凝胶渗透色谱仪(GPC,PL-GPC 50 Plus)上测试聚合物分子量及其分布,流动相为0.1 mol/L 的硝酸钠水溶液,标准样品为聚氧化乙烯水溶液。P(SBMA-GMA)共聚物在水中形成的胶束形貌用透射电子显微镜(TEM,JEM-1200EX)进行观察。在核磁共振仪(300 MHz,Varian Mercury Plus)进行核磁(H NMR)测试,样品溶于氘代试剂(D2O),浓度为10~20 mg/mL。
Tween20 的临界胶束聚集数由荧光光谱测试得到[10]。以芘(Py)饱和水溶液为溶剂,配制一系列浓度的Tween20 溶液,并加入一定量的荧光猝灭剂(十六烷基氯化吡啶,CPC),在LS/Perkin Elmer荧光光谱仪上进行测试。
2 结果与讨论
2.1 引发剂用量的影响
SBMA 与GMA 的自由基胶束共聚体系极为复杂,不仅包含水溶性单体SBMA 与疏水单体GMA的胶束共聚,同时还存在 SBMA 的溶液聚合和 GMA 的乳液聚合。本体系中引发剂用量对P(SBMA-GMA)共聚物的影响如表1所示,用质量分数10%的共聚物水溶液的表观粘度(ηa)间接表征共聚物分子量的大小。对于自由基聚合而言,聚合物的分子量一般随引发剂用量的增加而降低。由表1可知,本体系中只有当引发剂用量较高(0.20%)时,ηa才有较大幅度下降,即共聚物分子量降低。而引发剂用量相对于单体总量的摩尔分数为0.05%~0.15%时,共聚物水溶液ηa变化不大。此外,除了引发剂用量较低(0.05%)时共聚物中亲水单体(SBMA)含量较低,引发剂用量较高时,SBMA含量相对较高。结果表明,当引发剂用量为0.08%时,共聚物产率较高,亲水性单体SBMA 含量最高,得到的共聚物分子量较高,更有利于呈现超亲水、抗蛋白吸附的特性。

表1 引发剂用量对P(SBMA-GMA)性质的影响Table 1 Effect of the amount of initiator on properties of P(SBMA-GMA)
2.2 表面活性剂用量的影响
由于亲水单体SBMA 是一种两性聚电解质,对正负离子均较为敏感,故本体系选择非离子型表面活性剂Tween20,其作用主要是通过形成胶束增容疏水单体GMA,而与SBMA 相互作用较弱。GMA胶束是实现SBMA 和GMA 共聚的重要条件。实验考察了Tween20 用量对共聚物中亲水单体SBMA含量的影响,结果见表2。
体系中Tween20 用量远超过其临界胶束浓度(CMC为5.1×10-5mol/L)[10],即大部分Tween20分子呈胶束状态。表2所示,当GMA 用量较少时,共聚物中SBMA 含量随Tween20 用量的增加而下降。当GMA 用量较大时,Tween20 用量对共聚物中SBMA 含量影响变得不明显,仅稍有增大。相同Tween20 用量下,GMA 用量低时得到的共聚物中亲水单体SBMA 含量显然较高。当GMA 用量为2 g时,Tween20 用量较低意味着形成的胶束尺寸较大且数量少,胶束总表面积随之减小,此时共聚物中以SBMA 单体自聚为主,随着Tween20 用量增加,胶束尺寸减小但数量增加,此时更利于胶束内部的GMA 参加反应,在自由基寿命一定时,疏水单体GMA 含量增加,而SBMA 含量较低。当GMA 用量为4 g 时,Tween20 用量对共聚物中SBMA 含量影响变得不明显。当Tween20 用量较大时,共聚物中SBMA 含量稍有增大。这可能是因为Tween20 用量过高时,虽然胶束总表面积较大,但单个胶束尺寸很小,包括SBMA 齐聚物自由基在内的自由基难以进入小尺寸胶束引发GMA 单体聚合。
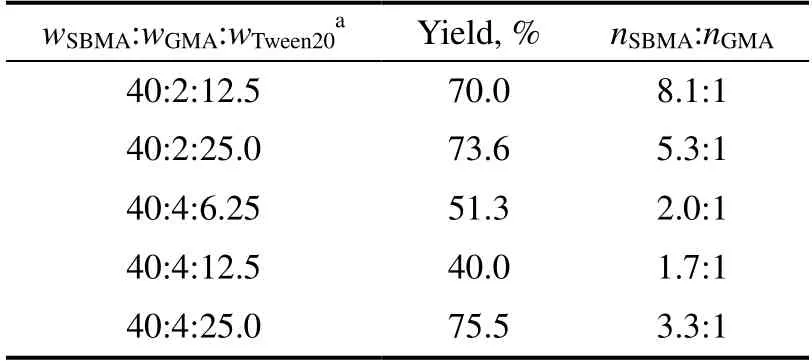
表2 Tween20 用量对P(SBMA-GMA)性质的影响Table 2 Effect of the amount of Tween20 on properties of P(SBMA-GMA) copolymer
2.3 搅拌速率的影响
搅拌速率影响单体与自由基以及自由基之间的碰撞几率,对胶束尺寸也有较大影响,进而影响最终产物的性质。实验考察了搅拌速率对P(SBMA-GMA)性质的影响,结果如表3所示。
由表3可知,搅拌速率增加,所得共聚物的分子量(ηa)和共聚物中SBMA 含量均逐渐下降,这可能是因为该反应体系中,引发剂浓度仅为单体的0.08%,分解产生的自由基被大量反应单体包围,自由基寿命较长。当搅拌速率较低时,被单体包裹的自由基不易发生双基终止,链终止速率下降,以亲水单体SBMA 的自聚为主,因此共聚物分子量及SBMA 含量均较大。此外值得注意的是,当转速为150 r/min 时,经提纯后的共聚物在水中的溶解性较差,而转速较高时(>250 r/min),得到的共聚物在水中溶解性较好。这与搅拌速率对胶束尺寸的影响有关,低速搅拌时,胶束数目少,单个胶束内部增容的GMA 较多,导致引入到共聚物分子链中的疏水嵌段长,共聚物在水中溶解性较差;而高速搅拌时,虽然产物中疏水单体所占比例稍有增加,但嵌段长度有所减小,共聚物的溶解性得到改善。

表3 搅拌速率对P(SBMA-GMA)性质的影响Table 3 Effect of the stirring rate on properties of P(SBMA-GMA)
2.4 P(SBMA-GMA)疏水嵌段共聚物的结构及性质
由以上可知,本研究通过自由基胶束聚合实现了亲水单体SBMA 和疏水单体GMA 的共聚。图1给出了SBMA 均聚物(PSBMA)和P(SBMA-GMA)的红外谱图。可以发现,共聚物特征基团的红外吸收峰均有体现,其谱峰归属如下[11]:2 963 cm-1归属于甲基的不对称伸缩振动峰;亚甲基的不对称伸缩振动峰和对称伸缩振动峰分别位于2 926,2 853 cm-1处;酯基的伸缩振动峰在1 730 cm-1有所体现;1 484 cm-1处的吸收峰属于C-N+基团的特征吸收峰;1 182,1 040 cm-1处的两个吸收峰归属于SO3—基团,即共聚物中的SBMA。环氧基团的对称伸缩振动出现在1 260 cm-1处,强度较弱,均聚物中无该峰,由此判定共聚物中存在GMA;环氧基团不对称伸缩振动吸收峰位于810~950 cm-1,由于GMA含量较低以及C-H 基团在960 cm-1处也有吸收峰,该吸收峰可能被覆盖,在红外谱图中较难体现。
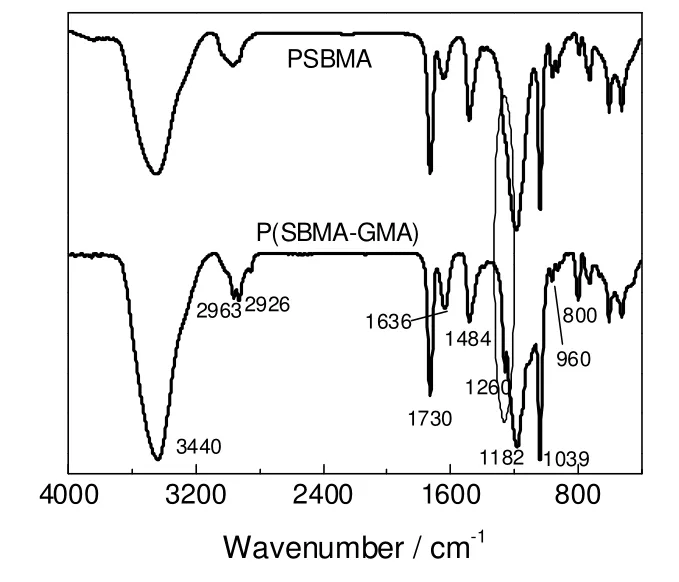
图1 PSBMA 和P(SBMA-GMA)的红外图谱Fig.1 FT-IR spectrum of PSBMA and P(SBMA-GMA) copolymer
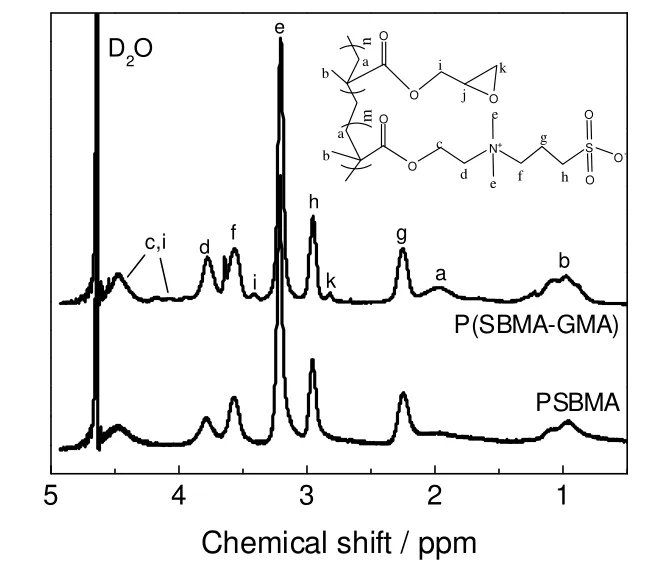
图2 P(SBMA)和P(SBMA-GMA)的H NMR 图谱Fig.2 H NMR spectrum of PSBMA and P(SBMA-GMA) copolymer
图2给出了PSBMA 和P(SBMA-GMA)的H NMR 谱图。比较均聚物和共聚物谱图,可以发现,共聚物的谱图比均聚物多了几个吸收峰:化学位移(δ)为2.82 ppm 的谱峰(k)是疏水单体GMA 中环氧基团的亚甲基上氢原子的吸收峰,由于共聚物中GMA 含量低,且环氧基团为疏水基团,主要分布于分子链的内部,被SBMA 的亲水基团包裹,故此吸收峰强度较弱,环氧基团上叔碳氢原子的吸收峰(j)出现在δ为3.41 ppm 处,δ为3.9~4.5 ppm 处的几个谱峰(c 和i)为与酯基相连的亚甲基上氢原子的吸收峰,较分散,强度较弱。H NMR 谱图进一步证明了自由基胶束共聚得到了疏水改性两亲性离子共聚物。
P(SBMA-GMA)共聚物中的疏水嵌段长度和数量可分别通过式(1)和式(2)计算得到[12]。

式中,[MH]为疏水单体的浓度,mol/L;Nagg为Tween20 的临界胶束聚集数;[Tween20]为Tween20的浓度;CMC为Tween20 的临界胶束浓度;NH为疏水嵌段长度;n为疏水嵌段数量;Mn为共聚物的数均分子量;wGMC为共聚物中GMA 的质量分数;MGMA为GMA 的摩尔质量。Nagg由荧光光谱测得[10]。反应温度50 ℃下,Tween20 在水中的Nagg为4。表4给出了不同单体投料比时共聚物大分子链中GMA疏水嵌段长度及嵌段数量。可以发现,单体投料比对疏水嵌段长度及数量均有显著影响。显然,GMA用量越高,疏水嵌段长度及数量均增大。相同单体投料比下,乳化剂用量越大,形成的GMA 胶束数量越多,共聚后形成的疏水嵌段长度越短,数量越多。因此,可以通过单体投料比及乳化剂用量来控制共聚物的微观结构。
表4同时给出了不同单体投料比得到的P(SBMA-GMA)共聚物的分子量及其分布。由于均聚物和共聚物水溶液中均存在静电相互作用,在GPC 测试过程中,需加入氯化钠屏蔽电荷间相互作用。从表4可以看到,疏水单体用量增加,对共聚物分子量及其分布无显著影响。相同疏水单体用量时,Tween20 用量增加,使共聚物分子量稍有降低,分布变宽。与一般自由基聚合共聚物的分子量低于均聚物分子量不同,相同条件下PSBMA 均聚物的分子量远低于共聚物,这是由于SBMA 单体之间的相互排斥作用不利于单体的聚集,导致不能形成高分子量的产物;而对于共聚物而言,GMA 胶束的存在削弱了SBMA 之间的静电排斥作用,同时,SBMA 齐聚物自由基也可进入胶束引发GMA 单体聚合,进而获得较高分子量的共聚物。
疏水嵌段在水中形成疏水缔合,使P(SBMA-GMA)大分子以胶束形式存在。图3是浓度为0.2%的P(SBMA-GMA)共聚物水溶液的TEM 照片,可以看到,P(SBMA-GMA)共聚物大分子确实呈胶束形态,且胶束尺寸分布较宽,最小的约为100 nm,而大的可达500~600 nm,大胶束又由小胶束缔合而成(见图中插图)。在用铜网制样过程中,随着溶剂水的挥发,胶束间发生聚集,形成较大的缔合体。此外,亲水单体SBMA 带正负异种电荷,存在静电相互吸引作用[13],也可形成较大缔合体。以上两个原因共同造成胶束尺寸分布较宽。

表4 P(SBMA-GMA)共聚物分子量分布Table 4 The molecular weight distribution of P(SBMA-GMA) copolymer

图3 P(SBMA-GMA)在水溶液中的透射电镜结果Fig.3 The TEM photo for P(SBMA-GMA) copolymer aqueous solution
3 结 论
采用自由基胶束共聚法成功合成了具有疏水微嵌段结构的聚两性电解质共聚物P(SBMA-GMA),考察了聚合反应条件对共聚物结构的影响,得到以下主要结论:
引发剂用量较高时,得到的共聚物中SBMA 的含量相对较高。当GMA 用量较少时,共聚物中SBMA 含量随Tween20 用量的增加而下降;当GMA 用量较大时,Tween20 用量对SBMA 含量影响不明显。搅拌速率增加,共聚物水溶液的ηa和共聚物中SBMA 含量均逐渐下降。GMA 用量越高,疏水嵌段长度及数量均增大。相同单体投料比下,乳化剂用量越大,形成的GMA 胶束数量越多,共聚后形成的疏水嵌段长度越短,数量越多。P(SBMA-GMA)大分子在水中以胶束形式存在。自由基胶束聚合得到的P(SBMA-GMA)共聚物的分子量高于SBMA 均聚物。
[1]Lacik I, Selb J, Candau F.Compositional heterogeneity effect in hydrophobically associating water-soluble polymers prepared by micellar copolymerization [J].Polymer, 1995, 36(16):3197-3211.
[2]Chen H, Wang Z M, Ye Z B, et al.The solution behavior of hydrophobically associating zwitterionic polymer in salt water [J].Journal of Applied Polymer Science, 2014, 39707(1-7).
[3]An H Y, Lu C G, Wang P X, et al.A novel hydrophobically associating polyampholytes of poly(AM/AA/AMQC12):preparation,characterization, and solution properties [J].Polymer Bulletin, 2011, 67(1):141-158
[4]Gamil M, Margaillan A, Martin I, et al.Synthesis ofN-aeryl- andN-arylakylacrylamide and micellar copolymerization with acrylamide [J].European Polymer Journal, 2000, 36(9):1853-1863.
[5]Akiyoshi K, Kang E C, Kurumada S, et al.Controlled associative of amphiphilic polymers in water:thermosensitive nanoparticles formed by self-assembly of hydrophobically modified pulluans and poly(N-isopropylacrylamides) [J].Macromolecules, 2000,33(9):3244-3249.
[6]Dobrynin A V, Colby R H, Rubinstein M.Polyampholytes [J].Journal of Polymer Science Part B:Polymer Physics, 2004,42(19):3513-3538.
[7]Wang D, Wu T, Wan X J, et al.Purely salt-responsive micelle formation and inversion based on a novel schizophrenic sulfobetaine block copolymer:structure and kinetics of micellization [J].Langmuir, 2007, 23(6):11866-11874.
[8]Mary P, Bendejacq D D.Interactions between sulfobetainebased polyzwitterions and polyelectrolytes [J].Journal Of Physical Chemistry B, 2008, 112(8):2299-2310.
[9]Hill A, Candau F, Selb J.Properties of hydrophobically associating polyacrylamides:influence of the method of synthesis [J].Macromolecules, 1993, 26(17):4521-4532.
[10]章苏宁, 张 健, 宋晓秋, 等.稳态荧光探针法测定Tween 系列非离子表面活性剂临界胶束浓度 [J].光谱实验室, 2010,27(4):1231-1236.Zhang Suning, Zhang Jian, Song Xiaoqiu, et al.Determination of critical micelle concentration(CMC) of nonionic surfactants by steady state fluorescence probe method [J].Chinese Journal of Spectroscopy Laboratory, 2010, 27(4):1231-1236.
[11]曾幸荣, 吴振耀, 侯有军, 等.高分子近代测试分析技术 [M].广州:华南理工大学出版社, 2007:80-105.
[12]Branham K D, Shafer G S, Hoyle C E, et al.Water-soluble copolymer:Microstructural investigation of pyrenesulfonamide-labeled polyelectrolytes - variation of label proximity utilizing micellar polymerization [J].Macromolecular.1995, 28(18):6175-6182.
[13]Singh S S, Siddhanta A K, Meena R, et al.Intermolecular complexation and phase separation in aqueous solutions of oppositely charged biopolymers [J].International Journal of Biological Macromolecules, 2007, 41(2):185-192.
猜你喜欢
原子与分子物理学报(2021年2期)2021-03-29 07:31:46
理化检验-化学分册(2020年12期)2021-01-26 00:41:40
吉林建筑大学学报(2018年1期)2018-03-05 02:21:37
化工进展(2015年3期)2015-11-11 09:18:44
材料研究与应用(2015年4期)2015-08-23 11:39:36
体育世界(学术版)(2015年3期)2015-07-01 17:15:41
西安建筑科技大学学报(自然科学版)(2014年2期)2014-11-12 13:04:38
应用化工(2014年11期)2014-08-16 15:59:13
应用化工(2014年9期)2014-08-10 14:05:08
采矿技术(2013年6期)2013-11-19 01:50:18
