
不对称催化亲电氟化反应研究进展
2015-11-17 12:10汪忠华巫辅龙吴范宏
应用技术学报 2015年1期
汪忠华, 巫辅龙, 吴范宏
(上海应用技术学院化学与环境工程学院,上海 201418)
不对称催化亲电氟化反应研究进展
汪忠华, 巫辅龙, 吴范宏
(上海应用技术学院化学与环境工程学院,上海 201418)
在有机氟化学领域中,α-氟代羰基化合物具有特异的生物活性,在有机合成中也可以作为合成砌块,其合成方法学的研究是目前研究的热点和难点之一.不对称亲电氟代反应是直接构建α-氟代羰基骨架的有效方法,主要用到的催化剂包括钌、铜、钪为催化中心的金属催化剂和奎宁、手性有机磷酸为主的有机小分子催化剂.介绍了最近催化对映体选择性亲电氟化反应领域研究情况.
不对称催化;对映体选择性亲电氟化反应;金属催化剂;有机催化剂
氟元素是地球上第十三大丰富的元素,但自然界中的含氟天然产物却很少,这可能是由于氟原子的强电负性和低的亲核性导致的.氟原子的引入,能诱导化合物的物理化学和生物特性,如生物活性、新陈代谢的稳定和药动力学的特性发生显著改变.氟原子的大小介于氢原子和氧原子之间[1],C—F键长也相似地介于C—H和C—O键之间,但C—F键能在三者中最强.因此,当分子中氢原子被氟原子取代后,其空间大小并不会有显著变化,但分子的电子云分布、偶极矩、脂溶性、稳定性等都有明显改变.
分子中引入氟的方法很多,不对称催化亲电氟化反应是其中之一,它是制备手性α-氟代羰基化合物的一种有效途径,α-氟代羰基结构常常在药物分子中出现.氟红霉素(Flurithromycin)是法玛西亚公司(Pharmacia)开发的一种新型大环内酯类呼吸道感染的主要致病菌肺炎链球菌抗生素[2].氟林卡那(Flindokalner,MaxiPost)是百时美施贵宝公司(Bristol Myers Squibb)开发的一种大钾离子通道开放剂[3].氟红霉素和氟林卡那的结构式见图1.

图1 具有生物活性的α-氟代羰基化合物Fig.1 Bio-activeα-fluoro carbonyl compounds
不对称催化亲电氟化反应主要通过手性催化剂作用,亲电氟代试剂作用于底物羰基的邻位并引入氟原子而完成.2000年,Hintermann等[4-5]报道了第一个对映选择性催化氟化反应,使用氟化试剂Selectfluor和催化量为5 mol%的手性四价钛配合物,对不同取代基的β-酮酸酯进行了对映选择性氟化反应研究,对映体选择性最好的为82%.2005年,Enders等[6]报道了第一例以(S)-脯氨酸衍生物为手性有机催化剂,Selectfluor为氟化试剂诱导的对映选择性氟化反应,研究了醛、酮的不对称氟化反应,但对映体过量率并不理想,只有34%,之后发现,氟化试剂的选择性对反应非常重要,同时必须抑制氟化产物的烯醇化.同样,2005年,Beeson等[7]研究发现了一种普遍能够对醛类衍生物氟代后得到很高的对映体选择性(ee值)的有机催化剂咪唑烷酮化合物.在23°C反应中N-氟代双苯磺酰胺(NFSI)为氟化试剂,催化量为2.5 mol%的咪唑烷酮化合物为有机催化剂,氟代后也能得到很高的ee值,可达到98%.随后,Mauro等[8]以NFSI为氟化试剂,采用不同手性催化剂对直链脂肪醛的不对称氟化反应进行了系统研究,筛选出高效催化剂咪唑烷酮化合物并应用于含有不同取代基的脂肪醛的氟化,得到较高对映选择性的α-氟代醛衍生物(86%~96%ee).Steiner等[9]同样也在2005年报道了对于不是很稳定的咪唑烷酮化合物催化生成苯乙醛,也能够以高收率和高对映体选择率得到相应的氟化产物,进而将醛基还原,可制备更稳定的、高光学纯度的2-氟代醇衍生物.
在不对称亲电氟代反应中,常用的亲电氟代试剂有Selectfluor、氟代吡啶、NFSI等[10-18],具体结构如图2所示.所用催化剂主要包括钌、铜、钪为催化中心的金属催化剂和奎宁、手性磷酸为主的有机小分子催化剂.本文报道自2009年以来,金属催化剂、有机金属催化剂和有机小分子催化剂对α-羰基化合物不对称亲电氟代反应的研究进展.

图2 常用亲电氟代试剂Fig.2 Common electrophilic fluorinating agents
1 金属催化剂
1.1 钌为催化中心
在有关文献报道中,有效的氟代试剂大多只有3种,分别为Selectfluor,N-fluoropyridinium和N-fluorobenzenesulfonimide(NFSI).2009年,Martin等[19]报道了以Ag HF2为氟代试剂,钌为催化剂的2-烷基苯基乙醛不对称氧化α-氟化反应如图3所示.反应过程中氟化试剂Ag HF22.4倍物质的量,催化剂[RuCl2(PNNP)]SbF6量为5 mol%,1,2-二氯乙烷为溶剂,反应温度为60°C,反应时间为24 h,对不同α-位上的苯基乙醛进行了研究,不同苯乙醛衍生物经过氟代反应后最高的收率可达35%,ee值为27%,见表1.
R的取代基位阻变大后,相应的产率和ee值也会变小,表明产物的反应受位阻的影响.虽然在反应过程中产率和ee值都比较低,但新氟化试剂的研究为今后研究和发展提供了更广阔的空间.
1.2 铜为催化中心
2009年,Assalit等[20]报道了以Cu(OTf)2与胺手性配体形成的复合物为催化剂,酮酯化合物在二氯甲烷中经NFSI作用的不对称亲电氟代反应(见图4),得到相对应产物6a、6b和6c的ee值和较好的收率,不同配体4和5b对应的产物产率和ee值分别为75%(9%ee)和78%(17%ee)、77%(27%ee)和60%(32%ee)、54%(18%ee)和55%(27%ee).手性配体5b和化合物4相比,不同反应底物6a、6b和6c对应产物的收率影响不大,但手性配体5b得到对应产物的ee值明显高于化合物4,这很可能取决于配体的空间位阻,如表2、图5所示.

图3 钌催化的不对称亲电氟化反应过程Fig.3 The reaction of asymmetric electrophilic fluorination with ruthenium catalyst

表1 钌催化的不对称亲电氟化反应Tab.1 The asymmetric electrophilic fluorination with ruthenium catalyst
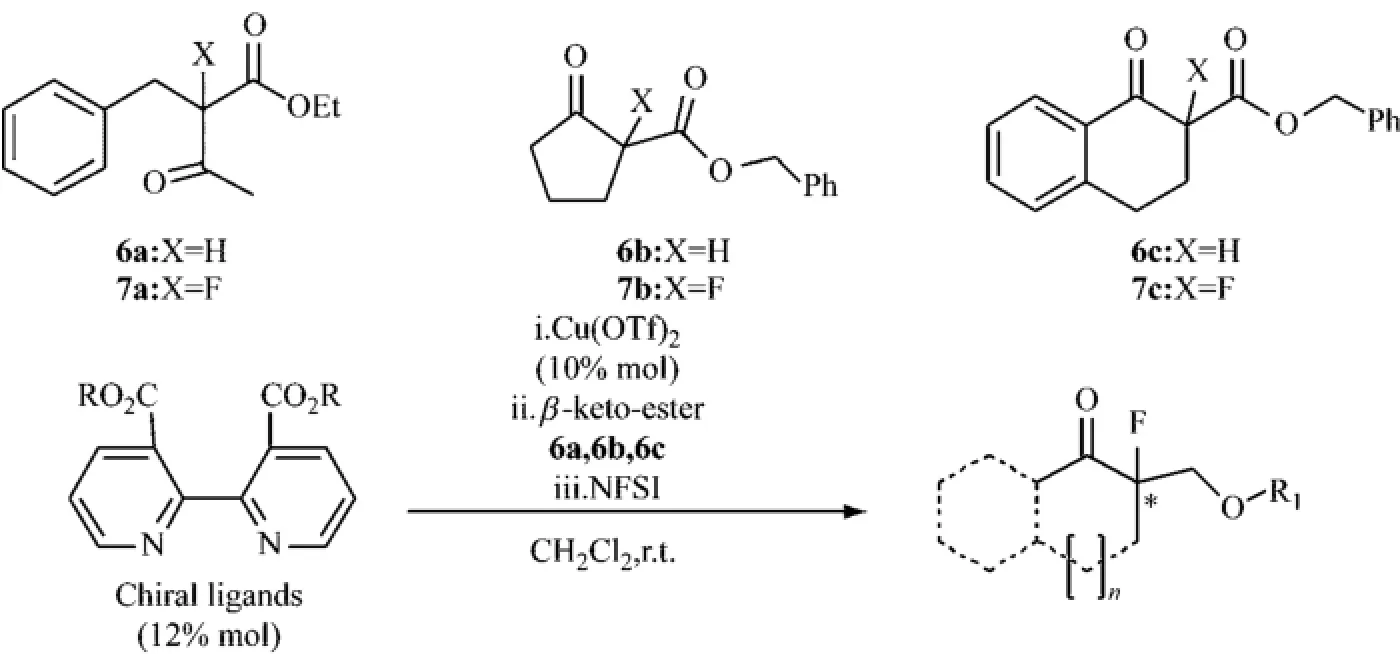
图4 铜催化的不对称亲电氟化反应过程Fig.4 The reaction of asymmetric electrophilic fluorination with copper catalyst

表2 铜催化的不对称亲电氟化反应Tab.2 The asymmetric electrophilic fluorination with copper catalyst

图5 手性胺配体结构式Fig.5 Structural formula of chiral ligand
除此之外,研究还发现,当以化合物5a为配体,化合物6b为反应底物时,氟化试剂NFSI与Selectfluor和N,N-difluoro-2,2’-bipyridinium bis-(tetrafluoroborate)相比,具有反应时间短和收率高的优势(分别对应的收率和时间为64%迅速反应,63%反应3 d和13 d后仍无反应),如表2所示.对不同金属催化剂的考察发现,二价铜的催化效果优于其他金属催化试剂(收率为77%,27%ee),如表3所示.
1.3 钪为催化剂的氟化反应
2012年,Li等[21]以一步法直接在吲哚酮五元环的N原子相对的C3位置上引入氟原子,完成氟代亲电反应,如图6所示.

表3 不同金属对亲电氟化反应的影响Tab.3 Effects of different metals on the electrophilic fluorination

图6 钪催化的不对称亲电氟化反应Fig.6 The asymmetric electrophilic fluorination with scandium catalyst
在反应过程中,Li等以NFSI(1.2 mol)作为氟化试剂,5 mol%~10 mol%的9-Sc(OTf)3(见图6)为催化剂,120 mol%Na2CO3为碱,CHCl3为溶剂,得到的收率最高达98%和99%ee.还研究了以化合物9为配体(见图7)的不同金属(如:Ni(ClO4)2· 6H2O、Mg(OTf)2、(OTf)3和La(OTf)3)催化剂对反应收率和ee的影响,结果表明,反应收率和ee普遍偏低.在5种金属催化剂中钪的催化效果最好,9f为配体时ee值最高可以到达88%(见图8和表4).这也同样体现出了不同金属配体化合物的几何构型的差异对对映体选择性的影响[22].当改变配体取代基的空间位阻后,发现小位阻的配体反而得到的ee有所下降(取代基位阻大小:9b>9c>9d>9e,对应的ee值分别为87%、75%、50%和13%,见表4).
化合物12为Bristol Meyer Squibb为治疗中风研发的氟化羟吲哚[23],其合成须经过3步反应.但在Li的报道中提出了一种一步合成化合物12(Maxipost)的方法,还对化合物12的合成进行了条件优化,以81%的收率和96%的ee值得到化合物12,如图9所示.

图7 胺手性配体9Fig.7 Chiral ligand 9

表4 不同金属对亲电氟化反应的影响Tab.4 Effects of different metals on the electrophilic fluorination

图8 不同金属的亲电氟化反应Fig.8 The asymmetric electrophilic fluorination with different metals catalyst

图9 化合物12的合成方法Fig.9 Synthetic methods of compound 12

图10 DHQB/Selectfluor组合的不对称亲电氟化体系Fig.10 Asymmetric electrophilic fluorination system of DHQB/Selectfluor
2 有机小分子催化剂
2.1 奎宁生物碱为催化剂
奎宁生物碱是一种作为不对称亲电氟代反应的主要催化剂,这主要是由于奎宁生物碱具有高效的催化效果.但在反应过程中,奎宁生物碱必须先和氟化试剂结合,才能和氟代底物发生反应得到较好的对映体选择性[24],如图10和表5所示.

表5 DHQB/Selectfluor组合的不对称亲电氟化体系结果Tab.5 Results of asymmetric electrophilic fluorination system of DHQB/Selectfluor
通过X-ray对DHQB的结构分析如图11所示,研究者认为晶体结构表现出来的DHQB其中的一部分(右下方)和另一部分(左上方)相比空间位阻比较大,故左上方部分能够容易地诱导来自氟试剂的一个氟原子.这样空间结构就像口袋一样,处在DHQB之间,能够形成对对映体选择性氟化转变的有利手性环境.金鸡纳碱DHQB和Selectfluor组合的氟代反应机理大致如图12所示.
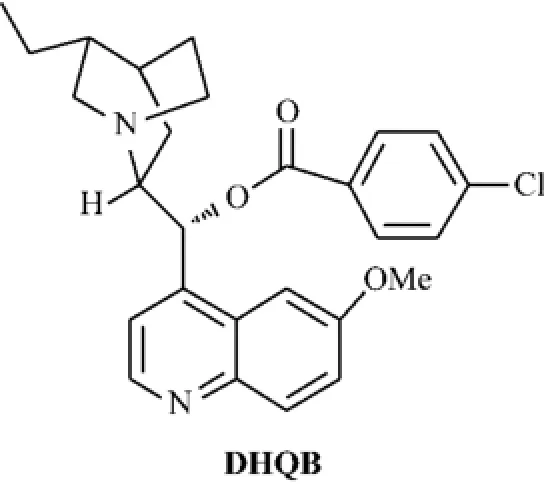
图11 金鸡纳碱DHQBFig.11 Cinchona alkaloid DHQB

图12 金鸡纳碱DHQB和Selectfluor组合的氟代反应机理Fig.12 The fluorination mechanism mediated by the cinchona alkaloid DHQB and Selectfluor
在过去大部分以奎宁生物碱为催化剂的亲电氟化反应中,使用的氟化试剂普遍为Selectfluor、N-fluoropyridinium和NFSI.2011年,Yasui等[25]报道了一种新的氟化试剂N-Fluoro-(3,5-di-tertbutyl-4-methoxy)benzenesulfonimide(NFBSI),如图13所示.
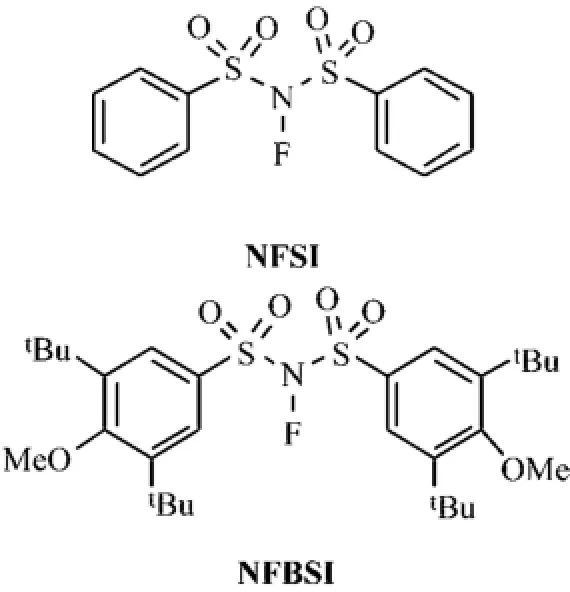
图13 氟化试剂NFSI和NFBSI的结构Fig.13 Fluorinating reagents of NFSI and NFBSI
NFBSI与NFSI相比位阻大大增加,但是当NFSBSI和NFSI在相同的反应条件下反应时,能够提高对映体的选择性,增加幅度可高达18%,比如:1.2倍物质的量的氟化试剂NFSI和NFBSI分别与硅醚衍生物15a和15b在10 mol%的奎宁生物碱催化剂,6.0倍物质的量的K2CO3,乙腈为溶剂,反应温度为0°C到室温,反应时间为2 d的情况下明显可见,NFBSF为氟化试剂对应的对映体的选择性明显高于NFSI,分别为70%ee和52%ee(16a),58% ee和40%ee(16b),高于18%(见图14),主要是由于NFBSI的空间位阻的增大,大大增加了其对对映体的选择性.
2.2 手性磷酸为催化剂
Hamashima等[26-29]以手性有机磷化合物为催化剂,开发了由羟基架桥的手性复核Pd(μ-OH)配合物17a(见图15),从而实现手性金属磷催化剂与氟化试剂发生氟化反应.

图14 NFBSI和NFSI为氟化试剂的氟化反应Fig.14 Fluorination with the reagents of NFBSI and NFSI

图15 手性有机磷配体Fig.15 Chiral phosphorus ligands
Phipps等[30]在2013年报道了纯粹的以(S-3,3’-双-2,4,4-三环己基-苯基)-[1,1’]-联萘-3,3’-二氧磷酸((S)-TCYP)为催化剂(见图16)也能够实现不对称亲电氟化反应(见图17),并且对映体选择性非常高,为今后的研究工作提供更好的研究思路和方法,尽可能摆脱有机金属配体对氟化反应的依赖.
在反应过程中加入碳酸钠可以加快反应速率,得到较高的对映体选择性.氟化试剂与碳酸钠反应能够形成具有活性的Selectfluor碳酸盐,该盐离子有可能诱导氟原子转移.

图16 (S-3,3’-双-2,4,4-三环己基-苯基)-[1,1’]-联萘-3,3’-二氧磷酸((S)-TCYP)Fig.16 (S)-3,3’-Bis-(2,4,4-tricyclohexyl-phenyl)-[1,1’]-binaphthaenyl-2,2’-dioxaphosphoric-acid

图17 手性磷酸作用的不对称亲电氟代反应Fig.17 The asymmetric electrophilic fluorination with chiral phosphoric acid catalyst
之后,Wu等[31]也在2013年报道了以(S-3,3’-双-2,4,4-三异丙基-苯基)-[1,1’]-联萘-3,3’-二氧磷酸((S)-TRIP)和(R)-6,6’-二(2,4,6-三异丙基苯)-1,1’-螺二茚-7,7’-二氧磷酸((R)-STRIP)为催化剂(见图18)的不对称氟化反应.报道中,以烯烃化合物20a和20b为底物(见图18和表6),Selectfluor(1.35 equiv.)为氟化试剂,Na2CO3(1.45 equiv.)为碱,催化剂为TRIP(10%mol),反应温度为10°C,经过18 h反应后,也可以得到很好的ee值,最高可达90%ee(见表6).

图18 烯烃化合物的不对称氟代反应Fig.18 The asymmetric electrophilic fluorination with olefin

表6 不同条件下的氟代反应情况Tab.6 Fluorination results under different conditions
由表6可见,不同溶剂和不同催化试剂对相同反应底物经过不对称氟化反应后得到的对映体也不尽相同.为此,通过以(R)-STRIP为催化剂改变酰胺衍生物氟代反应底物的结构作为研究,在甲苯为溶剂,Selectfluor(1.35 equiv.)为氟代试剂,Na2CO3(1.45 equiv.)为碱,室温反应18 h后,也可得到较好的对映体选择性的产物24,如图19和表7所示.

图19 不同底物的不对称亲电氟代反应Fig.19 Different substrates on asymmetry electrophilic fluorination
当改变R取代基时,对映体的选择性也会发生明显变化.当烷甲基数目增大,从1个甲基变成3个(序号1~3)时,空间位阻明显增大,相应地,对映体选择性也从30%ee优化到75%ee.其原因主要是空间位阻增大,当位阻增大时,空间位阻的效应明显大于电子效应(t-Bu>i-Pr>Me).当R取代基为二氟甲基时,对映体选择性为30%ee(序号5),这与甲基取代时一样,主要原因可能是氟原子构型和其空间旋转的共同作用,从而减小空间位阻.由表6(序号5、6)可知,氟原子的增加,对映体选择性就会明显增加(氟原子的取代数目由2个变成3个对应的对映体选择性分别为30%ee和82%ee).通过对比可以看到R为3个Cl取代基团的对映体选择性(93%ee)明显高于3个F原子的取代对映体选择性(82%ee),故空间位阻的影响明显大于吸电子效应(见表7,序号6、7).
从某种意义上,新的有机小分子作为不对称亲电氟化反应的催化剂的发现为今后研究提供了更加宽广的思路,摆脱金属有机配体作为催化剂的思维.

表7 不同底物对不对称亲电氟代反应的影响Tab.7 Effects of different substrates on asymmetry electrophilic fluorination
3 结 语
对映体选择性氟化反应仍然是研究的热门领域,这必将引起对不对称催化亲电氟代反应的关注和研究,从而不断促进氟化反应的发展和创新.特别是现今医药行业对含氟药物日益扩大需求,因此,合成具有生物活性新的氟代产物和研究其药用价值将具有更加重大的意义.
[1] Hagan D O.Understanding organofluorine chemistry. An introduction to the C—F bond[J].Chem Soc Rev,2008,37(2):308-319.
[2] Grassi G G,Alesina R,Bersani C,et al.In vitro activity of flurithromycin,a novel macrolide antibiotic[J].Chemioterapia,1986,5(3):177-184.
[3] Young B L,Cooks R G,Madden M C,et al.Chiral purity assay for Flindokalner using tandem mass spectrometry:Method development,validation,and benchmarking[J].J Pharmaceut Biomed Ana,2007,43(5):1602-1608.
[4] Hintermann L,Togni A.Catalytic enantioselective fluorination ofβ-ketoesters[J].Angew Chem Int Ed,2000,39(23):4359-4362.
[5] Hintermann L,Perseghini M,Togni A,et al.Titanium-catalyze stereoselective geminal heterodihalogenation ofβ-ketoesters[J].Org Lett,2003,5(10):1709-1712.
[6] Enders D,Hüttl M R M.Direct organocatalytic alpha-fluorination of aldehydes and ketones[J].Synlett,2005,6:991-993.
[7] Beeson T D,Mac Millan D W C.Enantioselective organocatalyticα-fluorination of aldehydes[J].J Am Chem Soc,2005,127(24):8826-8828.
[8] Mauro Marigo,Doris Fielenbach,Ala Braunton,et al.Enantioselective formation of stereogenic carbonfluorine centers by a simple catalytic method[J].Angew Chem Int Ed,2005,44:3703-3706.
[9] Steiner D D,Mase N,Barbas C F.Direct asymmetric α-fluorination of aldehydes[J].Angew Chem Int Ed,2005,44:3706-3710.
[10] Pihko P M.Enantioselectiveα-fluorination of carbonyl compounds:Organocatalysis or metal catalysis[J]. Angew Chem Int Ed,2006,45(4):544-547.
[11] Hamashima Y,Sodeoka M.Enantioselective fluorination reactions catalyzed by chiral palladium complexes[J].Synlett,2006,10:1467-1478.
[12] Prakash G K S,Beier P.Construction of asymmetric fluorinated carbon centers[J].Angew Chem Int Ed,2006,45(14):2172-2174.
[13] Bobbio C,Gouverneur V.Catalytic asymmetric fluorinations[J].Org Biomol Chem,2006,4(11):2065-2075.
[14] Shibata N,Ishimaru T,Nakamura S,et al.New approaches to enantioselective fluorination:Cinchona alkaloids combinations and chiral ligands?/metal complexes[J].Fluorine Chem,2007,128(5):469-483.
[15] Brunet V A,O’Hagan D.Catalytic asymmetric fluorination comes of age[J].Angew Chem Int Ed,2008,47(7):1179-1182.
[16] Ma J A,Cahard D.Update 1 of:Asymmetric fluorination,trifluoromethylation,and perfluoroalkylation reactions[J].Chem Rev,2008,108(9):1-43.
[17] Furuya T,Kuttruff C A,Ritter T,et al.Carbon-fluorine bond formation[J].Drug Discov Devel,2008,11(6):803-819.
[18] Ueda M,Kano T,Maruoka K.Organocatalyzed direct asymmetricα-halogenation of carbonyl compounds[J].Org Biomol Chem,2009,7(10):2005-2012.
[19] Martin A,Antonio T,Antonio M.Asymmetric oxidativeα-fluorination of 2-alkylphenylacetaldehydes with Ag HF2and ruthenium/PNNP catalysts[J].Science Direct,Journal of Fluorine Chemistry,2009,130(8):702-707.
[20] Assalit A,Billard T,Chambert S,et al.2,2’-Bipyridine-3,3’-dicarboxylic carbohydrate esters and amides.Synthesis and preliminary evaluation as ligands in Cu(II)-catalysed enantioselective electrophilic fluorination[J].Tetrahedron:Asymmetry,2009,20(5):593-601.
[21] Li Jun,Cai Yunfei,Chen Weiliang,et al.Highly enantioselective fluorination of unprotected 3-substituted oxindoles:One-step synthesis of BMS 204352(MaxiPost)[J].J Org Chem,2012,77(20):9148-9155.
[22] 姜永莉,刘兆鹏.对映选择性亲电氟化反应研究进展[J].有机化学,2009,29(9):1362-1370.
[23] Hewawasam P,Gribkoff V K,Pendri Y,et al.The synthesis and characterization of BMS-204352(Maxi-Post)and related 3-fluorooxindoles as openers of maxi-K potassium channels[J].Bioorg Med Chem Lett,2002,12(7):1023-1026.
[24] Shibata N,Suzuki E,Asahi T,et al.Enantioselective fluorination mediated by cinchona alkaloid derivatives/Selectfluor combinations:Reaction scope and structural information for N-fluorocinchona alkaloids[J].J Am Chem Soc,2001,123(29):7001-7009.
[25] Yasui H,Yamamoto T,Ishimaru T,et al.N-Fluoro-(3,5-di-tert-butyl-4-methoxy)benzenesulfonimide(NFBSI):A sterically demanding electrophilic fluorinating reagent for enantioselective fluorination[J]. Journal of Fluorine Chemistry,2011,132(3):222-225.
[26] Hamashima Y,Yagi K,Takano H,et al.An efficient enantioselective fluorination of variousβ-ketoesters catalyzed by chiral palladium complexes[J].J Am Chem Soc,2002,124(49):14530-14531.
[27] Hamashima Y,Suzuki T,Takano H,et al.Catalytic enantioselective fluorination of oxindoles[J].J Am Chem Soc,2005,127(29):10164-10165.
[28] Hamashima Y,Suzuki T,Shimura Y,et al.An efficient catalytic enantioselective fluorination ofβ-ketophosphonates using chiral palladium complexes[J]. Tetrahedron Lett,2005,9(46):1447-1450.
[29] Suzuki T,Goto T,Hamashima Y,et al.Enantioselective fluorination of tert-butoxycarbonyl lactones and lactams catalyzed by chiral Pd(II)-bisphosphine complexes[J].J Org Chem,2007,72(1):246-250.
[30] Phipps R J,Toste F D.Chiral anion phase-transfer catalysis applied to the direct enantioselective fluorinative dearomatization of phenols[J].J Am Chem Soc,2013,135(4):1268-1271.
[31] Wu J,Wang Yiming,Drljevic A,et al.A combination of directing groups and chiral anion phase-transfer catalysis for enantioselective fluorination of alkenes[J].Proc Natl Acad Sci,2013,110(34):13729-13733.
(编辑 吕丹)
Research Progress of Asymmetric Catalytic Electrophlic Fluorination
WANG Zhong-hua, WU Fu-long, WU Fan-hong
(School of Chemical and Environmental Engineering,Shanghai Institute of Technology,Shanghai 201418,China)
In the field of organic fluorine chemistry,α-fluoro carbonyl derivatives possess specific biological activity and can be used as synthetic building block in organic synthesis.The exploration of its synthetic methodology has been one of the hot and difficult spots in current scientific research.Asymmetric electrophilic fluorination is an effective method for directly constructingα-fluoro carbonyl skeleton,which mainly employs metal catalysts(ruthenium,copper,scandium)and organocatalysts(quinine,chiral phosphate).Based on the different catalysts,the recent progress in catalytic enantioselective electrophilic fluorination was introduced.
asymmetric catalysis;enantioselective electrophilic fluorination;metal catalyst;organocatalyst
O 622
A
1671-7333(2015)01-0009-10
10.3969/j.issn.1671-7333.2015.01.002
2014-08-23
中国科学院有机氟化学重点实验室研究基金资助项目(ZX2003-06);上海市高校青年教师培养基金资助项目(ZZyyy13002);上海应用技术学院引进人才基金资助项目(YJ2013-04)
汪忠华(1982-),男,讲师,博士,主要研究方向为有机氟化学、药物合成化学.E-mail:zhonghua_wang@sit.edu.cn
猜你喜欢
材料工程(2022年3期)2022-03-20
井冈山大学学报(自然科学版)(2021年3期)2021-09-10
无机化学学报(2020年7期)2020-07-20
中成药(2017年9期)2017-12-19
科技创新导报(2016年30期)2017-03-15
中国卫生标准管理(2015年17期)2016-01-20
中国当代医药(2015年9期)2015-03-01
医药导报(2015年6期)2015-02-10
中国塑料(2014年12期)2014-10-17
郑州大学学报(理学版)(2013年2期)2013-03-11
