
分子对接研究手性共价有机框架材料的对映体识别能力
2021-09-10 00:35张显显张忠杰
井冈山大学学报(自然科学版) 2021年3期
黄 露,张显显,张忠杰
分子对接研究手性共价有机框架材料的对映体识别能力
*黄 露,张显显,张忠杰
(闽江学院海洋学院化学工程系,福建,福州 350108)
基于分子对接方法,使用AutoDock软件计算手性共价有机框架材料CCOF5与手性对映体相互作用的结合能。计算结果表明:CCOF5与对映体的结合能差大小与对映体分离度大小相一致;CCOF5与对映体的结合能大小与对映体出峰时间顺序基本一致。采用Avogadro软件对AutoDock分子对接构象进行分析,并对手性识别机理进行探讨。
分子对接;手性共价有机框架材料;对映体;手性识别
共价有机框架材料(COFs)是由有机构筑单元通过共价键形成的多孔晶体聚合物材料[1]。由于其具有高的孔隙率、功能化的内表面以及可调节的结构特点,COFs在能量存储[2]、光电器件[3]、传感[4]等领域有着广阔的应用前景。近年来随着人们对手性化合物研究的不断深入,手性已经成为化学、生物、材料等学科的研究热点,手性COFs也因此应运而生。作为一种新型的手性分离材料,手性COFs已被成功用于液相色谱[5]、气相色谱[6]、毛细管电色谱[7]拆分手性对映体。然而由于手性COFs材料出现时间较短,尚未有关于其手性识别机理的报道。
在过去近30年里,科研工作者对色谱拆分对映体进行了大量的分子模拟研究,旨在从分子水平上研究分子之间的弱相互作用以及结合能,从而使实验结果合理化、可预测化,并进一步阐释手性识别机理。以环糊精为例,人们采用分子力学[8]、分子动力学[9]等分子模拟方法研究其与客体的包合作用及手性识别机理。与上述分子模拟方法相比,广泛应用于药物开发的分子对接是一种更有效的计算方法[10]。对接过程根据几何和化学互补性模拟大分子与小分子的结合构象和取向,从而确定特定的相互作用。然后通过评估大分子与小分子之间的相互作用能来预测得分函数,进而对相互作用进行量化。目前,分子对接已被成功用于研究环糊精[11]、纤维素[12]等手性材料与手性对映体之间的相互作用。CCOF5是一种新型的手性COFs,已被成功用于液相色谱拆分手性对映体[5]。本研究使用AutoDock进行分子对接模拟,系统研究了CCOF5对四种外消旋化合物(即1-苯基-2-丙醇、1-苯基-1-戊醇、1-苯基-1-丙醇、1-(4-溴苯基)乙醇)的对映体识别能力。采用Avogadro软件对CCOF5与手性对映体的结合构象进行分析,并进一步对CCOF5识别手性对映体的机理进行探索。
1 原理和方法
1.1 受体CCOF5和配体小分子的结构优化
CCOF5的结构以及色谱拆分对映体的数据均来自相关文献[5]。CCOF5和四种外消旋化合物的化学结构如图1所示。为了简化计算,选取CCOF5基本构筑单元中易于发生相互作用的手性结构部分作为受体分子(如图1虚线框内结构)。使用Orca软件在B3LYP-D3/def2-TZVP计算水平上对受体CCOF5及配体分子(四对对映体)进行结构优化。

A. (±)-1-苯基-1-戊醇;B. (±)-1-苯基-1-丙醇;C. (±)-1-苯基-2-丙醇;D. (±)-1-(4-溴苯基)乙醇
1.2 分子对接模拟
使用自动对接程序AutoDock 4.2.6对CCOF5和四对对映体分别进行分子对接。采用拉马克遗传算法结合基于网格的能量评估方法,根据受体、配体分中所有原子类型的原子间相互作用势,计算格点盒子(盒子大小为15 Å × 22.5 Å × 15 Å,格点间距为0.375 Å)。共进行100次独立对接,其他参数使用默认值。考虑到能量越低越稳定以及结合位点等因素,确定CCOF5与对映体结合最有可能的构象,记录其结合能并保存构象用于后续分析。
2 结果与讨论
2.1 分子对接结果
按照步骤1.1和1.2进行分子对接,所得到的CCOF5与四种外消旋化合物的结合能及结合能差列于表1(表中选择性因子及分离度数据来自文献5)。从表1可以看出,对映体间结合能差绝对值由大到小:1-苯基-2-丙醇>1-(4-溴苯基)乙醇>1-苯基-1-丙醇>1-苯基-1-戊醇;对映体的分离度由大到小:1-苯基-2-丙醇>1-(4-溴苯基)乙醇>1-苯基-1-丙醇>1-苯基-1-戊醇;对映体的手性选择性因子由大到小:1-苯基-1-丙醇>1-苯基-1-戊醇>1-苯基-2-丙醇>1-(4-溴苯基)乙醇。上述结果表明,CCOF5与对映体的结合能差大小与分离度大小相一致,即对映体的结合能差越大,对映体的分离度也越大;然而结合能差大小与选择性因子大小不存在一致性。
文献[5]所分离的对映体为外消旋化合物,并未对具体对映体进行确认。通常结合能越低,相互作用越强,其出峰时间越迟。因此,从结合能大小上可以推断:R型对映体出峰时间应当早于S型对映体出峰时间。根据文献[5],1-苯基-1-戊醇两个对映体的出峰时间约为20 min和24 min,1-苯基-1-丙醇相应约为25 min和30 min,1-苯基-2-丙醇相应约为24 min和28 min,1-(4-溴苯基)乙醇约为26 min和31 min。如表1所示,CCOF5与S型对映体的结合能绝对值由大到小:1-(4-溴苯基)乙醇(31 min)>1-苯基-1-丙醇(30 min)>1-苯基-2-丙醇(28 min)>1-苯基-1-戊醇(24 min)(括号内为出峰时间)。该结果与S型对映体的时间排序一致。CCOF5与R型对映体的结合能绝对值由大到小:1-(4-溴苯基)乙醇(26 min)>1-苯基-1-丙醇(25 min)>1-苯基-1-戊醇(20 min)>1-苯基-2-丙醇(24 min)(括号内为出峰时间)。除了1-苯基-1-戊醇,该结果与R型对映体的时间排序基本一致。
表1 CCOF5与四种外消旋化合物的结合能、结合能差、选择性因子及分离度
Table 1 Binding energy, binding energy difference, selectivity factor and resolution of CCOF5 with four racemates
外消旋物结合能(ΔE)kJ/moL结合能差(ΔΔE)keal /mol选择性因子分离度Rs S-enantiomerR-enantiomer (±)-1-苯基-1-戊醇- 2.92- 2.900.021.200.92 (±)-1-苯基-1-丙醇- 3.01- 2.980.031.211.26 (±)-1-苯基-2-丙醇- 2.93- 2.870.061.191.52 (±)-1-(4-溴苯基)乙醇- 3.41- 3.360.051.171.44
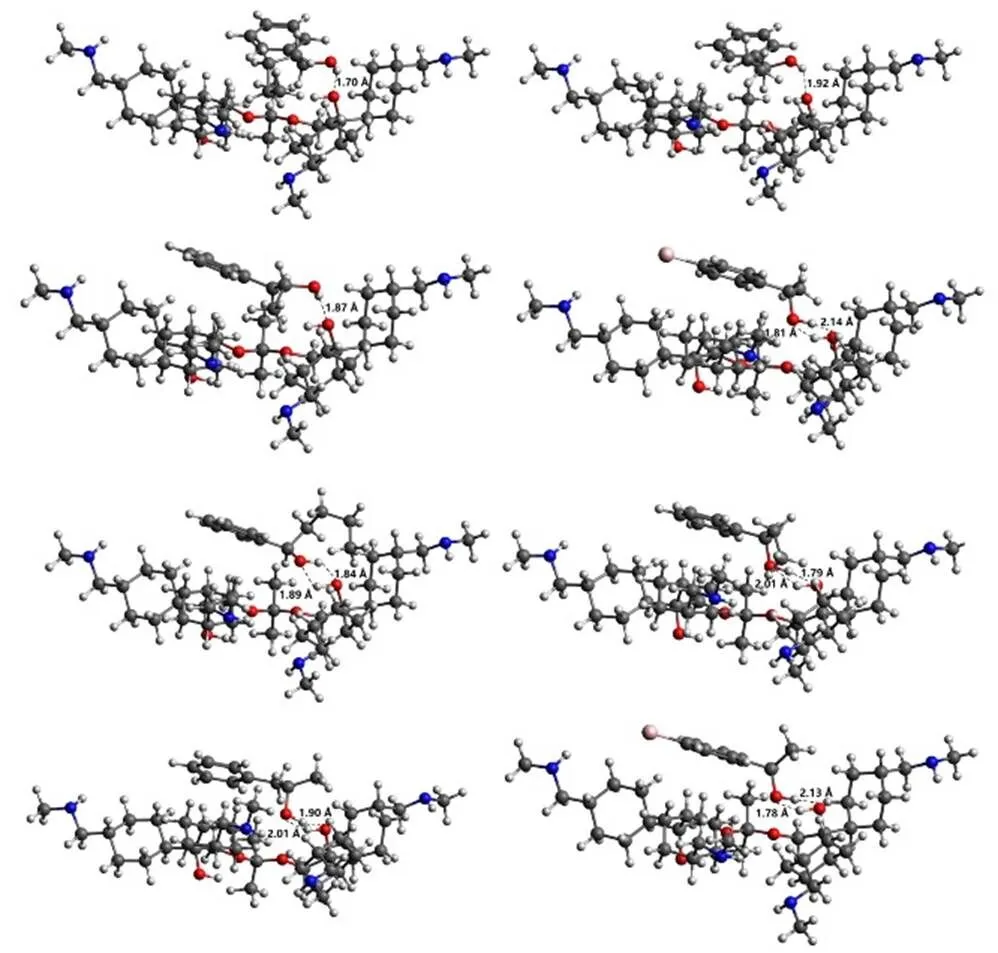
从左到右依次为(±)-1-苯基-1-戊醇,(±)-1-苯基-1-丙醇,(±)-1-苯基-2-丙醇,(±)-1-(4-溴苯基)乙醇,上面为S型对映体,下面为R型对映体
2.2 对映体识别机理
使用Avogadro软件对AutoDock分子对接构象进行分析。CCOF5与四种外消旋化合物对接结果如图2所示。从图中可以看出,CCOF5发生相互作用的部位在其手性碳原子连接的羟基上,而四种外消旋化合物发生相互作用的部位亦在其手性碳原子连接的羟基上。CCOF5与四种外消旋化合物之间均存在O-H...O氢键相互作用。由于外消旋化合物R型与S型空间构象不同,CCOF5与R型对映体之间可以形成两个O-H...O氢键,而与S型对映体间只形成一个O-H...O氢键。只有1-(4-溴苯基)乙醇是例外,其R型和S型对映体都可以与CCOF5形成两个O-H...O氢键。这表明,除了1-(4-溴苯基)乙醇,其他三种外消旋化合物与R型对映体之间的氢键相互作用更强。然而,从表1可知,CCOF5与S型对映体的结合能更低,即CCOF5与S型对映体的相互作用更强,其结合更稳定。这也说明尽管CCOF5与R型对映体具有更强的氢键相互作用,但是由于其他分子间作用力(诸如范德华力、空间位阻效应等)的影响,CCOF5与R型对映体的相互作用力比S型对映体弱。
3 小结
本研究基于分子对接方法采用AutoDock软件计算CCOF5与四种外消旋化合物的结合能。计算结果表明,CCOF5与对映体的结合能差大小与分离度大小相一致,然而结合能差大小与选择性因子大小不存在一致性。CCOF5与S型对映体的结合能大小与对映体出峰时间顺序相一致,CCOF5与R型对映体的结合能大小与对映体出峰时间顺序基本一致。从结合能差大小,可以推断S型对映体出峰时间迟于R型对映体。另外,采用Avogadro软件对AutoDock分子对接构象进行分析,CCOF5与R型对映体之间可以形成两个O-H...O氢键,而与S型对映体之间只形成一个O-H...O氢键,S型1-(4-溴苯基)乙醇是例外,它可以与CCOF5形成两个O-H...O氢键。研究结果表明,分子对接软件AutoDock可用于模拟手性共价有机框架材料与手性对映体的相互作用过程,使用方便,效率更高,适用于研究手性共价有机框架材料的手性识别机理。
[1] 李霞,张赛男,高佳,等. 手性共价有机框架材料的研究进展与应用[J]. 中国科学:化学,2019, 49(5): 10-19.
[2] XU F, YANG S, CHEN X, et al. Energy-storage covalent organicframeworks:improvingperformance via engineering polysulfide chains on walls [J]. Chemical science, 2019, 10(23): 6001-6006.
[3] CAO L, GUO B, YU Y, et al. Two-dimensional covalent organic framework–graphene photodetectors: Insight into the relationship between the microscopic interfacial structure and performance [J]. ACS omega, 2019, 4(20): 18780-18786.
[4] SUN Y, XU L, WATERHOUSE G I, et al.Novel three-dimensional electrochemical sensor with dual signal amplification based on MoS2nanosheets and high-conductive NH2-MWCNT@ COF for sulfamerazine determination [J]. Sensors and Actuators B: Chemical, 2019, 281: 107-114.
[5] HAN X, HUANG J, YUAN C, et al. Chiral 3D covalent organic frameworks for high performance liquid chromatographic enantioseparation [J]. Journal of the American Chemical Society, 2018, 140(3): 892-895.
[6] QIAN H-L, YANG C-X, YAN X-P. Bottom-up synthesis of chiral covalent organic frameworks and their bound capillaries for chiral separation [J]. Nature communications, 2016, 7: 12104.
[7] WANG Y, ZHUO S, HOU J, et al. Construction of β-cyclodextrin covalent organic framework modified chiral stationary phase for chiral separation [J]. ACS Applied Materials & Interfaces, 2019, 11(51): 48363-48369.
[8] ALVIRA E. Influence of solvent polarity on the separation of leucine enantiomers by β-cyclodextrin: A molecular mechanics and dynamics simulation [J]. Tetrahedron: Asymmetry, 2017, 28(10): 1414-1422.
[9] CHEN Y, ZUO Z, DAI X, et al. Gas-phase complexation of α-/β-cyclodextrin with amino acids studied by ion mobility-mass spectrometry and molecular dynamics simulations [J]. Talanta, 2018, 186: 1-7.
[10] 刘志强,王博龙.基于TCMSP抗肿瘤中药小分子EGFR-TKI的研究 [J]. 井冈山大学学报:自然科学版, 2018, 39(2): 87-92.
[11] ZHANG C-L, LIU J-C, YANG W-B, et al. Experimental and molecular docking investigations on the inclusion mechanism of the complex of phloridzin and hydroxypropyl - β - cyclodextrin [J]. Food chemistry, 2017, 215: 124-128.
[12] ZHU B, ZHAO F, YU J, et al. Chiral separation and a molecular modeling study of eight azole antifungals on the cellulose tris (3,5-dichlorophenylcarbamate) chiral stationary phase [J]. New Journal of Chemistry, 2018, 42(16): 13421-13429.
MOLECULAR DOCKING STUDY OF THE ENANTIOMER ECOGNITION ABILITY OF CHIRAL COVALENT ORGANIC FRAMEWORKS
*HUANG Lu, ZHANG Xian-xian, ZHANG Zhong-jie
(Department of Chemical Engineering, Ocean College, Minjiang University, Fuzhou, Fujian 350108, China)
Based on the molecular docking method, the binding energy of chiral covalent organic framework (CCOF5) with chiral enantiomers was calculated by AutoDock software. The results showed that the binding energy differences between CCOF5 and enantiomers were consistent with the resolutions of enantiomers. The binding energy between them was consistent with the peak time sequence of enantiomers. Avogadro software was used to analyze the molecular docking conformation of AutoDock, and the chiral recognition mechanism was further explored.
molecular docking; chiral covalent organic frameworks; enantiomer; chiral recognition
O639
A
10.3669/j.issn.1674-8085.2021.03.004
1674-8085(2021)03-0016-04
2020-12-10;
2021-02-06
国家大学生创新创业训练计划项目(103952018023);福建省自然科学基金项目(2017J01418)
*黄 露(1983-),女,江西景德镇人,教授,博士,主要从事手性分离分析研究(Email: luhuang@mju.edu.cn);
张显显(1995-),女,河南商丘人,闽江学院海洋学院应用化学专业2016级本科生(E-mail:1287973517@qq.com);
张忠杰(1997-),男,福建宁德人,闽江学院海洋学院应用化学专业2016级本科生(E-mail:chiech@vip.qq.com).
猜你喜欢
分子催化(2022年1期)2022-11-02
分子催化(2022年1期)2022-11-02
科学导报(2022年41期)2022-07-13
湖南大学学报·自然科学版(2020年2期)2020-04-17
宇航材料工艺(2020年1期)2020-03-26
新民周刊(2018年8期)2018-03-02
饮食科学(2017年12期)2018-01-02
江苏农业科学(2017年20期)2017-11-30
少儿科学周刊·少年版(2015年1期)2015-07-07
湖北农业科学(2014年9期)2014-08-08
