
高效液相色谱—串联质谱法测定紫苏籽油中11种酚类化合物
2015-11-03 07:54吴娆等
分析化学 2015年10期
吴娆等


摘 要 基于新型富集材料磁性羧基化多壁碳纳米管与高效液相色谱-串联质谱,建立了紫苏籽油中11种酚类化合物同时分离与检测的方法。对影响富集分离效果的重要因素,如流动相、流动相中酸的添加量、流速、进样量及碰撞能量等条件进行了详细优化。在优化条件下,11种酚类化合物线性关系良好,相关系数为0.993~0.999,检出限(LOD, S/N=3)和定量限(LOQ,S/N=10)分别在0.02~0.70 ng/mL,0.06~2.0 ng/mL范围内,以50和500 ng/mL添加浓度水平进行方法学验证,回收率为79.6%~121.5%,相对标准偏差为0.06%~13.2%。将本方法应用于紫苏籽油样品的分析,其含量在7.32~68.6 ng/g之间。结果表明,本方法操作简单、准确可靠,适用于紫苏籽油等植物油中酚类化合物定量分析。
关键词 紫苏籽油; 酚类化合物; 磁固相萃取法; 高效液相色谱-串联质谱
1 引 言
紫苏(Perillafrutescen(L)Brit.)又名赤苏、香苏等,为唇形科一年生草本植物,是我国传统的药食两用植物。紫苏籽油是由紫苏籽经烘焙、压榨和精炼获得食用油[1],具有防止动脉粥样硬化、降血脂、抗氧化、抗衰老、抑菌、抗肿瘤、平喘等作用[2~7]。然而,其作用的分子机理尚不清楚[7]。目前紫苏籽油化学成分认识仅限于脂肪酸(主要α-亚麻酸[8]),挥发物[6],紫苏籽和果渣中迷迭香酸、芹黄素和木犀草素等[9]。酚类化合物具有抗氧化作用,预防心血管疾病,抑菌、抗癌和解毒护肝[10~13]。因此,建立一种灵敏的紫苏籽油中酚类成分同时测定方法对紫苏籽油功能解析与评价具有重要意义。
目前,关于多酚类化合物的检测已有很多报道,陈磊等[14]采用高效液相色谱法检测黄酒中多酚类成分;吕海涛等[15]以乙酸乙酯为提取剂,建立了苹果中酚类成分定量分析方法;张甜等[16]采用固相萃取-高效液相色谱测定烟草样品中多酚;胡秋芬等[17]采用微柱液相色谱法测定金银花中的多酚类化合物。由于植物油中多酚类物质含量底,样品基质复杂,酚类化合物分析需对样品进行富集和纯化[18],因此建立有效的前处理技术尤为重要。目前多采用液液萃取、固相萃取等技术,存在费时、费力、有机溶剂量大等缺点[19],多壁碳纳米管因其独特的物理结构,巨大的比表面积而备受关注[20~22]。作为固相萃取吸附剂,多壁碳纳米管具有优异的富集性能,并且通过对其表面进行羧基化修饰和磁性化,使得材料更加稳定,不易聚集,能够均匀分散在基质样品中,实现酚类物质的高选择性纯化富集,操作过程快速简单,仅需外加磁场即可分离[23]。
本研究以磁性羧基化多壁碳纳米管材料为基础,建立了植物油中天然酚类组分富集-高效液相色谱串联质谱分析方法,优化了如流动相、流动相中酸的添加量、流速、进样量及碰撞能量等条件,并应用于紫苏籽油中多酚成分的测定,为全面认识紫苏籽油中酚类组分,开展紫苏籽油的品质与营养功能评价提供了必要基础。
2 实验部分
2.1 仪器与试剂
液相色谱质谱TSQ三重四级杆(美国Thermo公司);HQ-60旋涡混合器(北方同正生物技术发展公司);BF-2000型氮气吹干仪(北京八方世纪科技有限公司);Milli-Q超纯水仪(美国Millipore公司);KQ-600B型超声波清洗器(昆山市超声仪器有限公司)。
紫苏籽油:从主产区收集代表性紫苏籽,采用实验室物理压榨方式获得紫苏籽油。
对羟基乙醇、肉桂酸、3,4-二羟基苯甲酸、对香豆酸、香草酸、咖啡酸、阿魏酸、丁香酸、芹黄素、木犀草素、槲皮素、甲酸(Sigma公司);甲醇(色谱纯,Merck公司);醋酸(优级纯,国药集团化学试剂有限公司)、FeCl3·6H2O(分析纯,国药集团化学试剂有限公司);石油醚、乙二醇、无水乙醇(分析纯,西陇化工股份有限公司);乙酸钠(分析纯,天津市德恩化学试剂有限公司);正己烷(HPLC纯,安徽时联特种溶剂股份有限公司);羧基化多壁碳纳米管(南京先丰纳米材料科技有限公司,长度约30 μm, 直径<8 nm, COOH含量: 3.86%);实验用水为Milli-Q 超纯水,其它试剂为分析纯。
2.2 HPLC-MS/MS分析条件
2.2.1 液相色谱条件 Hypersil Gold C18液相色谱柱(100 mm×2.1 mm i.d., 3 μm),柱温:30℃;流速:0.2 mL/min;进样量:10 μL;流动相: A为0.01%(V/V) 醋酸-甲醇溶液,B为0.01%(V/V)醋酸-水溶液;流动相洗脱程序见表1。
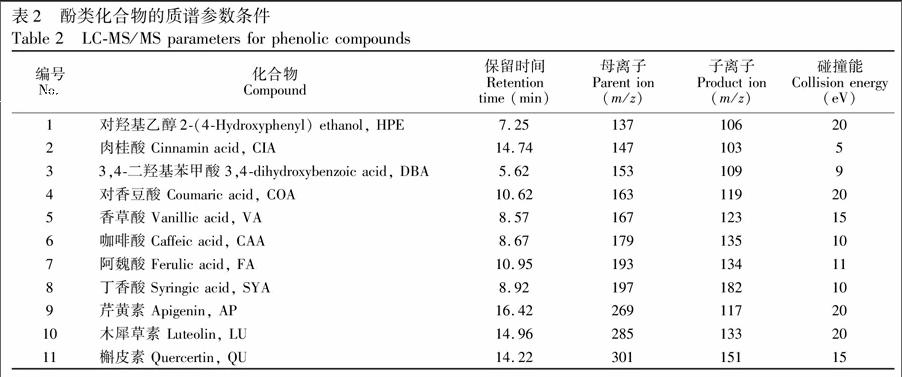
2.2.2 质谱条件 质谱条件:电喷雾离子源(ESI);多反应监测(MRM);负离子模式扫描;毛细管电压:3 kV;毛细管温度:270℃;气帘气(N2):30 units;辅助气(N2): 5 units;碰撞气(Ar): 1.5 mTorr;扫描时间:0.1 s;酚类化合物的质谱参数条件见表2。
2.3 混合标准溶液的配制
准确称取标准品对羟基乙醇、肉桂酸、3,4-二羟基苯甲酸、对香豆酸、香草酸、咖啡酸、阿魏酸、丁香酸、芹黄素、木犀草素、槲皮素各10.0 mg,分别用甲醇溶解,定容至10.0 mL,即得1.0 mg/mL单一标准品储备液,4℃避光保存。分别移取0.1 mL单一标准品混合,制备10.0 μg/mL混合标准贮备液,使用时用甲醇稀释成不同浓度梯度的系列标准工作液。
2.4 实验步骤
准确称取0.50 g紫苏籽油样于10 mL试管中,加入4.0 mL正己烷,增加样品流动性,使目标物更快地由样品转移到萃取剂中。充分混匀后,加入5.0 mg磁性纳米材料,涡旋提取2 min后,在试管外壁加磁铁,静置10 s,待液体呈透明状,将液体倒出。 加入1.0 mL石油醚淋洗杂质,涡旋15 s,再次在试管外壁加磁铁,静置10s,待液体透明,将液体倒出。选择乙醇为洗脱溶剂,加入3.0 mL 0.02%(V/V)甲酸-乙醇,超声6 min,过0.22 μm有机膜,氮吹浓缩,200 μL初始流动相比例定容,进样10 μL进行LC-MS/MS检测。
3 结果与讨论
3.1 流动相的选择
在高效液相色谱条件中流动相的选择至关重要。考察了甲醇-水、乙腈-水作为流动相的分离效果。结果表明,11种酚类化合物在甲醇-水体系获得较好分离,通过调整梯度洗脱程序,峰形和分离度基本达到要求。因此,本研究选择甲醇-水体系为流动相。针对部分目标物存在峰拖尾现象,加入少量酸调节溶液pH值,可使单个酚类的电离受到抑制,以中性分子的形态存在,极性减弱,增强在固定相上的保留[11]。
3.2 酸添加量优化
反相色谱柱填料表面由于存在残余的羟基,具有一定的酸性,而部分酚类化合物具有弱酸性,能够与残余硅羟基发生强的静电作用力,产生次级保留效应,洗脱被推迟,产生拖尾现象。由于酸性环境能够抑制硅羟基的电离,流动相中添加少量酸,可改善峰形,为了获得准确的定量分析结果,考察了酸添加量的影响。
采用醋酸调节流动相的酸性。对比了在水相中加入0.5%, 0.2%, 0.1%, 0.05%和0.01%(V/V)醋酸的出峰情况发现,在水相中加入0.01%(V/V)醋酸,效果最好,响应值最高; 利用同样的浓度梯度优化了甲醇相中醋酸添加量,结果发现,同样在0.01%(V/V)的效果最好。可能的原因是:有机相和水相中加入相同浓度的醋酸,保证了在梯度洗脱过程中,两种流动相混合时,保持恒定的醋酸浓度,pH值不发生变化,同时,这样可使基线更加平稳,容易平衡梯度,降低目标物在分离过程中受干扰因素。因此,流动相为0.01% (V/V)醋酸-甲醇和0.01%(V/V)醋酸。
3.3 流速和进样量选择
流速和进样量对提高目标化合物的分离效果非常重要。当流速为200 μL/min的效果优于250 μL/min; 考察了进样量分别为5, 10和20 μL的分离效果。当进样量为10 μL,目标物出峰情况良好,未出现前沿峰和拖尾峰。因此,本研究的流速选定200 μL/min,进样量为10 μL。
3.4 质谱条件优化
选取负离子模式监测,以\[M-H\]为母离子,选取离子丰度最强的碎片为子离子,通过优化锥孔电压、碰撞能量等参数,信号强度明显改善。质谱条件见表2。采用C18色谱柱,以0.01%醋酸-甲醇和0.01% 醋酸溶液进行梯度洗脱(梯度洗脱程序见表1),11种酚类化合物在30 min内可实现良好分离,保留时间见表2,目标物混合标准溶液的SRM色谱图见图1。
3.5 方法评价
配制10,50,100,200,500和1000 ng/mL的混合标准溶液,在优化的HPLC-MS/MS条件下进样检测,每个浓度重复进样3次,以目标物平均峰面积(Y)对标准溶液浓度(x, ng/mL)进行线性回归。根据信噪比(S/N)3倍和10倍分别确定目标物的检出限(LOD)和定量限(LOQ),结果见表3。11种酚类化合物都有较宽的线性范围,在各相应的线性范围内,线性关系良好(R2≥0.993);LOD为0.02~0.70 ng/mL,LOQ为0.07~2.00 ng/mL;准确度和精密度结果见表4,2个加标水平的回收率为79.61%~121.52%,RSD<15%,可以满足实际样品的测定需要。
将本方法与前期报道中食用油中酚类化合物定量分析方法相比,从表5可见,本方法分析时间短,线性范围宽,检出限低,能满足实际样品测定的需要。
3.6 实际样品分析
采用本方法对紫苏籽油实际样品进行检测,每个样品重复测定3次,检测结果见图2,11种酚类化合物含量分别为:对羟基乙醇8.00 ng/g、肉桂酸19.0 ng/g、3,4-二羟基苯甲酸7.32 ng/g、对香豆酸46.5 ng/g、香草酸68.6 ng/g、咖啡酸32.8 ng/g、阿魏酸55.7 ng/g、丁香酸22.4 ng/g、芹黄素43.3 ng/g、木犀草素7.96 ng/g和槲皮素13.4 ng/g;其中,香草酸、阿魏酸、对香豆酸、芹黄素、咖啡酸等含量较高,是紫苏籽油主要的潜在营养成分。
4 结 论
通过对流动相、流动相中酸的添加量、流速、进样量及质谱条件的优化和考察,建立了基于磁固相萃取技术LC-MS/MS检测紫苏籽油种11种酚类化合物的分析方法。本方法前处理操作简单,环境友好,实现了高灵敏度、高分析速度、低成本的目标,结果稳定、准确,为全面认识紫苏籽油中酚类组成,开展紫苏籽油的品质、营养功能评价提供了必要基础。
References
1 Park M H, Seol N G, Chang P S, Yoon S H, Lee J H. J. Food Sci., 2011, 76: C808-C816
2 Sadi A M, Toda T, Oku H, Hokama S. Exp. Anim., 1996, 45: 55-62
3 Kim H K, Choi H. Life Sci., 2005, 77: 1293-1306
4 Onogi N, Okuno M, Komaki C, Moriwaki H, Kawamori T, Tanaka T, Mori H, Muto Y. Carcinogenesis, 1996, 17: 1291-1296
5 Deng Y, Xie Q, Zhang S, Yao H, Zhang H. Planta Med., 2007, 73: 53-58
6 Kim S J, Yoon H N, Rhee J S. J. Am. Oil Chem. Soc., 2000, 77: 451-456
7 Lee S, Lee Y J, Sung J S, Shin H S. J. Korean Soc. Appl. Biol. Chem., 2015, 3: 325-334
8 Jung D M, Yoon S H, Jung M Y. J. Food Sci., 2012, 77: 1249-1255
9 Zheng G, Li S, Lin Z, Yang R, Zhao Y, Liu J, Yang S, Chen A. Phytochem. Anal., 2014, 114: 1007-1015
10 Zhao G, Qin G W, Wang J, Chu W J, Guo L H. Neurochem. Int., 2010, 56: 168-176
11 Lee J H, Park K H, Lee M H, Kim H T, Seo W D, Kim J Y, Baek I Y, Jang D S, Ha T J. Food Chem., 2013, 136: 843-852
12 Zhao G, Yao-Yue C, Qin G W, Guo, L H. Neurobiol. Aging, 2012, 33: 176-186
13 Yang S Y, Hong C O, Lee G P, Kim C T, Lee K W. Food Chem. Toxicol., 2013, 55: 92-99
14 CHEN Lei, HUANG Xue-Song. Chinese J. Anal. Chem., 2010, 38(1): 133-137
陈 磊, 黄雪松. 分析化学, 2010, 38(1): 133-137
15 L Hai-Tao, SUN Hai-Feng, QU Bao-Han, DAI Hong-Yi. Chinese J. Anal. Chem., 2007, 35(10): 1425-1429
吕海涛, 孙海峰, 曲宝涵, 戴洪义. 分析化学, 2007, 35(10): 1425-1429
16 ZHANG Tian, DONG Xue-Chang, WU Fang-Ping, YANG Guang-Yu, QIAO Yong-Feng. Chinese J. Anal. Chem., 2005, 33(3): 359-362
张 甜, 董学畅, 吴方评, 杨光宇, 乔永峰. 分析化学, 2005, 33(3): 359-362
17 HU Qiu-Fen, YANG Guang-Yu, HUANG Zhang-Jie, LI Hai-Tao, YIN Jia-Yuan. Chinese J. Anal. Chem., 2005, 33(1): 69-72
胡秋芬, 杨光宇, 黄章杰, 李海涛, 尹家元. 分析化学, 2005, 33(1): 69-72
18 XING Han-Zhu, WANG Xia, CHEN Xiang-Feng, WANG Ming-Lin, ZHAO Ru-Song. Chinese J. Anal. Chem., 2015, 43(3): 409-413
邢寒竹, 王 霞, 陈相峰, 王明林, 赵汝松. 分析化学, 2015, 43(3): 409-413
19 Wang W X, Yang T J, Li Z G, Jong T T, Lee M R. Anal. Chim. Acta, 2011, 690: 221-227
20 Soylak M, Ercan O. J. Hazard.Mater., 2009, 168(2): 1527-1531
21 Zhao Q, Wei F, Luo Y B, Ding J, Xiao N, Feng Y Q. J. Agr. Food Chem., 2011, 59(24): 12794-12800
22 KOU Li-Juan, LIANG Rong-Ning. Chinese Journal of Chromatography, 2014, 33(8): 817-821
寇立娟, 梁荣宁. 色谱, 2014, 33(8): 817-821
23 Meng L, Fu C, Lu Q. Prog. Nat. Sci., 2009, 19(7): 801-810
24 LUO Fan, FEI Xue-Qian, FANG Xue-Zhi, WANG Ya-Ping, WANG Jin-Yuan. Journal of Instrumental Analysis, 2011, 30(6): 696-700
罗 凡, 费学谦, 方学智, 王亚萍, 王金元. 分析测试学报, 2011, 30(6): 696-700
25 Ballus C A, Quirantes-Piné R, Bakhouche A, da Silva L F D O, de Oliveira A F, Coutinho E F, da Croce D M, Segura-Carretero A, Godoy H T. Food Chem., 2015, 170: 366-377
26 Herchi W, Sawalha S, Arrez-Romn D, Boukhchina S, Segura-Carretero A, Kallel H, Fernndez-Gutierrez A. Food Chem., 2011, 126(1): 332-338
27 Franco M N, Galeano-Díaz T, López , Fernndez-Bolaos J G, Snchez J, De Miguel C, Gil M V, Martín-Vertedor D. Food Chem., 2014, 163: 289-298
猜你喜欢
中国中药杂志(2016年21期)2017-02-16
中国中药杂志(2016年21期)2017-02-16
分析化学(2017年1期)2017-02-06
热带农业科学(2016年10期)2016-12-12
分析化学(2016年7期)2016-12-08
科学与财富(2016年28期)2016-10-14
分析化学(2015年9期)2015-09-11
分析化学(2015年1期)2015-01-20
