
制备条件对氧化铁催化合成碳酸二甲酯反应性能的影响
2015-10-24 09:09:08陈红萍刘广环梁英华赵新强
天然气化工—C1化学与化工 2015年1期
陈红萍,刘广环,梁英华,赵新强
(1.河北联合大学化学工程学院,河北 唐山 063000;2.河北工业大学化工学院,天津 300130)
由于铁具有不饱和的f轨道和可变价的特点,因此氧化铁在催化剂研究中得到了广泛关注,如用于乙苯脱氢[1]、催化湿式氧化[2]和脱硫[3]等反应中。氧化铁的制备条件对其理化性质的影响显著,通过不同方法可以得到不同粒度和形貌的氧化铁。但大多文献仅考察了其形貌和性质[4-5],仅有部分给出了其制备条件对特定使用情况的理化性质的分析。纳米氧化铁在尿素醇解制备碳酸二甲酯(DMC)反应中表现出较好的催化活性[6]。由于制备方法对其催化活性影响较大,在之前的CO2与甲醇直接合成DMC的反应中虽表现出一定的催化活性,但活性较低[7-9]。本文采用沉淀法制备氧化铁,重点考察了沉淀剂、溶液中Fe3+浓度、陈化时间、焙烧温度等对其形貌和催化合成DMC活性的影响。
1 实验部分
1.1 催化剂的制备
配制一定浓度的沉淀剂(NH3·H2O、KOH、K2CO3和 Na2CO3)水溶液和 Fe(NO3)3·9H2O 水溶液,采用并流滴入法制备催化剂,于70℃恒温水浴中磁力搅拌,溶液pH值控制在8~9之间。滴加完毕后常温陈化数小时,过滤,洗涤,用80℃恒温鼓风干燥,在马弗炉中于空气气氛下焙烧。
1.2 催化剂的表征
采用HCT-3微机差热天平对焙烧前的氧化铁前躯体进行热重/差热分析(TG/DTA),在空气气氛下程序升温,升温速率10℃/min,温度范围20℃~600℃。采用D/MAX2500PCX射线衍射仪对样品粉末进行XRD分析,X射线源为CuKα1,管电压为30kV,管电流为 40mA,扫描范围为 20°~70°。 采用S-4800型场发射扫描电子显微镜对样品进行形貌扫描,冷场发射电子源,加速电压0.5 kV~30 kV。N2吸附实验在Gemini V3365/2380全自动快速比表面积分析仪上进行,于300℃脱气3h,-197℃下吸附,对样品进行多点BET比表面积测试分析。采用AVATAR360型傅里叶红外光谱仪进行样品红外吸收光谱分析,分辨率0.5cm-1,波数精度0.01cm-1。
1.3 催化剂的活性评价
催化剂的活性评价在带有磁力搅拌和控温装置的SLP-4100四联平行反应釜中进行 (每个釜的有效体积为100mL)。典型操作如下:准确称取催化剂1g和甲醇494mmol,加入到反应釜中,用CO2置换釜内气体,用增压泵向釜内充入约6MPa(室温)的 CO2气体,搅拌转速 550r/min,加热至150℃,反应时间24h后,停止加热搅拌,自然冷却至室温。排出残余气体,对所得液相产物进行气相色谱分析。
1.4 反应产物分析
液体产物分析在GC7900气相色谱仪上进行。色谱柱为Ф0.53mm×30m×0.25μm石英毛细管柱,固定液为OV-101,氮气作载气,流速为17mL/min,柱箱温度70℃,汽化室温度为180℃,FID检测器,检测器温度180℃,进样量0.25μL,采用不分流进样。用正丁醇作内标物进行定量分析。
DMC的收率以单位质量催化剂所得DMC的物质的量来计,计算式如式(1)。
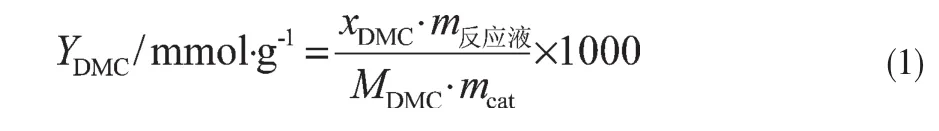
式中:xDMC-溶液中DMC的质量分数;m反应液-所得反应液的总质量,g;MDMC-DMC的相对分子质量;mcat-催化剂的质量,g。
2 结果与讨论
2.1 催化剂的表征
2.1.1 TG/DTA分析
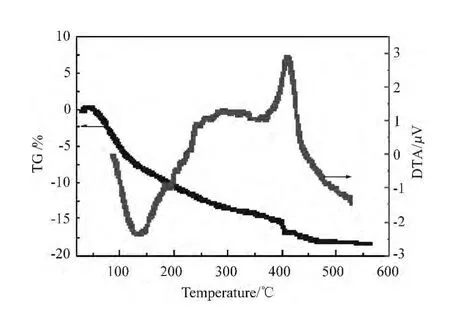
图1 催化剂前躯体的TG/DTA分析
为了确定焙烧温度对催化剂组成及晶型的影响,对以氨水为沉淀剂所制备的沉淀物经干燥后进行TG/DTA分析,结果如图1所示。氢氧化铁的脱水分解并没有明确的温度,而是在加热过程中的连续脱水过程。从图1可知,根据TG曲线的斜率不同,催化剂前躯体在升温过程中大致有3个失重阶段:第一失重阶段发生在27℃~192℃,对应为吸热失重,质量损失率为10.05%,可能为失去吸附水和第一个结合水过程[10];第二失重阶段发生在192℃~356℃,对应热效应为先吸热再放热,吸热过程产生的峰不明显,可能为失去第二个结合水的过程,质量损失率为1.3%,放热过程可能为氧化铁由无定形向晶体转化的过程;第三阶段失重发生在356℃~561℃,为失去部分第三个结合水过程,且在400℃左右有明显的强放热失重过程,可能为残存的硝酸根分解所致,质量损失率为3.47%。在561℃以上,仍有轻微的失重产生。计算从27℃~561℃质量损失率为18.23%,计算氢氧化铁总结合水质量分数为25.27%,说明氢氧化铁在561℃焙烧条件下仍有部分羟基存在。综合考虑不同焙烧温度可能产生氧化铁晶型和所结合羟基量的不同,本研究采用的焙烧温度分别是 250℃,300℃,350℃,400℃和 500℃,结合SEM、XRD考察不同焙烧温度所得催化剂的形貌性质和催化活性。
2.1.2 FTIR分析
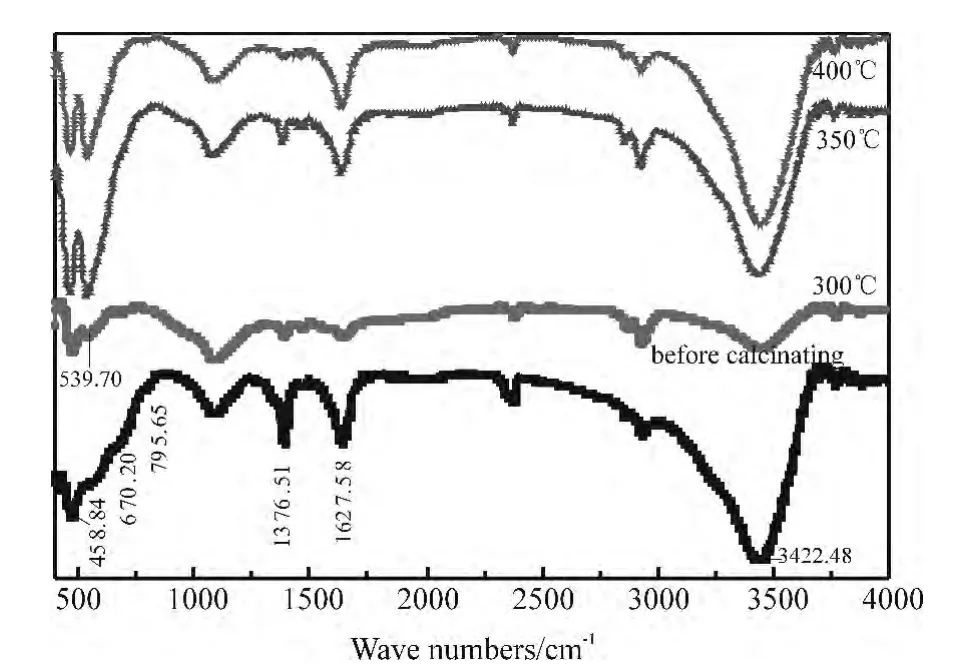
图2 不同焙烧温度所得催化剂的IR谱图
为了分析焙烧前后铁氧结合形式的变化,对催化剂焙烧前后试样进行红外光谱分析,结果如图2所示。波数为3422.58cm-1和1628.50cm-1处的吸收峰分别为-OH的伸缩振动和弯曲振动峰,应为样品中的O-H键和吸附水引起的。波数为1382.51cm-1处的吸收峰为硝酸根离子的特征吸收峰,比较焙烧前后和不同焙烧温度的谱图可见,当温度低于350℃时,该吸收峰均较高,当焙烧温度为400℃时,该吸收峰降低非常多,说明残存的硝酸根离子在350℃~400℃大部分分解,与TG/DTA分析结果一致 。 波 数 为 465.55cm-1、539.70cm-1、670.20cm-1、795.65cm-1处的吸收峰为Fe-O的特征吸收峰[11],其中波数为465.55cm-1、539.70cm-1处的吸收峰分别为八面体和八面体与四面体的变形振动吸收峰[12],波数为 670.20cm-1、795.65cm-1处的吸收峰为 FeOOH的吸收峰[13],与焙烧前相比,焙烧后催化剂539.70cm-1处吸收峰逐渐增强,波数为670.20cm-1、795.65cm-1处的吸收峰在焙烧温度在350℃时彻底消失,结合2.1.3的XRD分析,说明焙烧温度在350℃以上催化剂主要以单一的α-Fe2O3晶型存在。
2.1.3 XRD分析
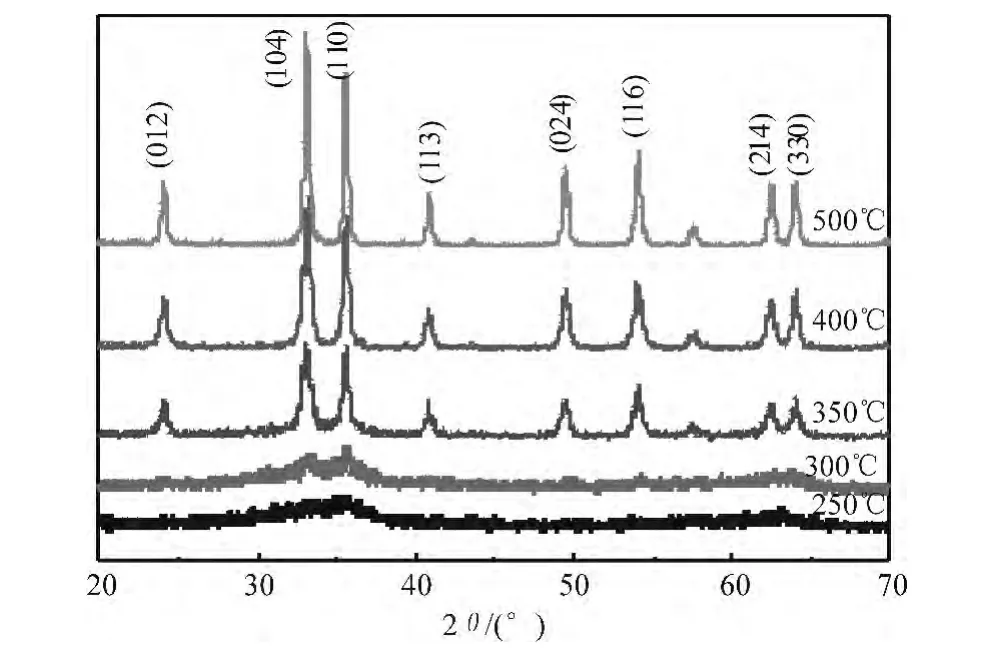
图3 不同焙烧温度所得催化剂的XRD谱图
为了考察焙烧温度对催化剂晶型的影响,对不同焙烧温度的催化剂进行XRD表征,结果如图3所示。由图可见,当焙烧温度低于250℃时,XRD衍射峰很弱且很宽,样品基本以无定型状态存在;当焙烧温度为300℃时,可以看到较明显的衍射峰,但峰仍旧很弱,说明所形成的晶型不完整且晶体颗粒直径很小;当焙烧温度为350℃时,衍射峰强度增强且峰宽变窄,衍射峰特点与赤铁矿的α-Fe2O3的XRD标准谱图一致,说明Fe2O3以三方菱心的α-Fe2O3晶型存在[14],这与FTIR分析结果一致。当焙烧温度继续升高,Fe2O3仍以α-Fe2O3晶型存在,但所有衍射峰强度均有增加,峰宽变窄,说明随着焙烧温度的升高,晶粒长大。
2.1.4 BET分析
对于非均相催化剂而言,催化剂的比表面积直接影响暴露的活性位数量,同时也对反应物和产物的传质阻力产生影响。催化剂制备条件对BET比表面积影响非常大(见图4)。由图4可知,催化剂的比表面积随着溶液中Fe3+浓度和焙烧温度的升高而下降,而随陈化时间的变化则存在一最佳值。分析其原因为,Fe3+浓度影响的是形成沉淀的过饱和度,由于Fe3+浓度低时,过饱和度较小,滴定沉淀所需时间较长,控制同样pH值所需氨水量较多,较稀的溶液形成的晶粒较小,同样陈化时间晶粒长大程度有限,体相与表层焙烧分解速率相当,不容易发生团聚,因此比表面积较大。随着溶液中Fe3+浓度的增加,过饱和度增大,形成沉淀速度加快,形成一次晶粒增多,同时伴有小晶粒溶解,大晶粒长大的过程,导致同样陈化时间和焙烧温度下催化剂BET比表面积减小。而陈化时间主要影响沉淀在母液中晶粒分布的变化,由于粒子直径大小不同,表面饱和蒸汽压不同,小粒子饱和蒸汽压大,导致小粒子溶解扩散沉积到较大粒子表层[15],随着陈化时间的延长,粒子直径分布趋于均匀,同样焙烧温度下比表面积增加,但继续增加陈化时间,会使粒子的直径进一步增大,导致比表面积反而降低。焙烧温度影响的不只是粒径,还有催化剂组成的变化,从曲线看在300℃~400℃之间,比表面积呈直线下降,结合IR和TG/DTA结果分析,低于400℃主要是氢氧化铁脱水较快的阶段,氢氧化铁的分解失水表现为比表面积急剧减小,当形成较稳定的氧化铁后,比表面积的降低主要表现为颗粒的团聚烧结,即颗粒的变大,从以下SEM表征结果可以得到进一步证实。

图4 催化剂制备条件对BET比表面积的影响
2.1.5 SEM分析
2.1.5.1 不同沉淀剂所得催化剂的SEM分析
不同沉淀剂所得催化剂的形貌不同。图5为不同沉淀剂在350℃焙烧温度下所得催化剂的SEM图。由图可见,K2CO3、Na2CO3为沉淀剂所制备的氧化铁颗粒粒径较大且分布不均匀,多有大块板结实体;氨水和KOH为沉淀剂所制备的催化剂粒径较小,颗粒直径均在100nm以内,但KOH为沉淀剂所制备的颗粒粘附团聚现象较严重,且颗粒粘附较紧密。氨水做沉淀剂所得催化剂颗粒分布较均匀,颗粒以松散形式衔接,且较KOH为沉淀剂所制备催化剂的颗粒直径要小。对样品表面进行EDS分析发现,用KOH、K2CO3和Na2CO3为沉淀剂所制备的氧化铁中均残存有沉淀剂的阳离子。说明以金属阳离子沉淀剂所得沉淀物中会残存有洗不去的对应的金属阳离子,而以氨水作沉淀剂,可以得到较纯净的沉淀物,结合催化剂活性评价可知,沉淀剂带入金属阳离子使催化活性降低。

图5 不同沉淀剂制备的Fe2O3的SEM-EDS谱图
2.1.5.2不同制备条件所得催化剂的SEM分析
制备条件对催化剂的形貌及颗粒粒径的影响较大。图6给出了以氨水为沉淀剂,不同Fe3+浓度、陈化时间、焙烧温度所得Fe2O3的SEM谱图。图6(A)为不同Fe3+浓度所得催化剂的SEM图,从图中可以看出,Fe3+浓度较小时,催化剂颗粒直径较小,且外形不规则,有颗粒粘附,随着溶液中Fe3+离子浓度的增加,颗粒直径变大,颗粒外形逐渐明晰。结合N2吸附/脱附结果分析可知,低Fe3+浓度所得小粒径催化剂可获得高比表面积。图6(B)为不同陈化时间所得催化剂的SEM图,从图中可以看出,陈化时间较短时,所得催化剂颗粒直径约在20nm~30nm之间,但有大块集结,且颗粒边界不清楚,呈模糊的雾状;当陈化时间为12h时,催化剂颗粒均匀,颗粒直径仍在20nm~30nm范围;当陈化时间为24h时,颗粒直径增至约50nm,为较规整的球形;当陈化时间增至36h时,颗粒直径进一步增加,且有大块团聚产生。颗粒直径的增加和团聚导致BET比表面积变小,与N2吸附/脱附分析结果一致。由图6(C)可以看出,焙烧温度越高所得催化剂的颗粒边界线越明显,由300℃焙烧所得催化剂颗粒的粘附集结到500℃焙烧所得催化剂颗粒的离散性分散。结合N2吸附/脱附和TG/DTA结果分析可知,催化剂BET比表面积随焙烧温度的升高而减小,并不完全是颗粒直径的影响,从300℃到350℃焙烧所得催化剂粒径看,由看似大块的颗粒团聚到形成较小纳米颗粒的堆积,比表面积反而减小,说明表观颗粒大小不是影响BET比表面积的主要因素,而催化剂本身组成变化才是主要原因,即由氢氧化铁逐渐分解脱水转化成氧化铁。400℃焙烧时,发生纳米颗粒的团聚和部分烧结,500℃焙烧时,烧结的纳米颗粒呈规整的外形颗粒,看似花生壳形,其BET比表面积均有不同程度的减小,说明350℃以上BET比表面积变小的原因是颗粒直径增大和颗粒团聚烧结所致。

图6 不同制备条件所得催化剂的SEM谱图
2.2 催化剂的反应性能研究
2.2.1 不同沉淀剂对所制备催化剂催化活性的影响
将不同沉淀剂所制备Fe2O3催化剂用于CO2与甲醇直接合成DMC反应体系中,考察不同沉淀剂所制备的催化剂对DMC收率的影响,结果见图7(a)。由图可知,催化剂的催化活性顺序为:氨水>KOH>K2CO3>Na2CO3,且以氨水为沉淀剂所制备的催化剂的催化活性远高于其他沉淀剂。结合不同沉淀剂的SEM-EDS表征结果,分析其原因可能为氨水沉淀剂所得催化剂颗粒均匀,且粒径小,同时氨水做沉淀剂所制备的催化剂不含有其他杂质阳离子,而其他3种沉淀剂制备的Fe2O3催化剂中会或多或少残留了K+、Na+离子杂质(见图5),从而使催化剂催化活性下降,可以判定少量碱金属离子的存在会使活性急剧下降。
2.2.2 溶液中Fe3+浓度对DMC收率的影响
以氨水为沉淀剂,考察溶液中不同Fe3+浓度所制备的催化剂的催化活性,结果见图7(b)。由图可见,随着溶液浓度的升高,催化剂的DMC收率呈现先增大后减小的趋势,即在溶液中Fe3+浓度为0.625mol/L时,DMC的收率出现了峰值。结合BET比表面积和SEM结果分析其原因为,当溶液浓度较稀虽有较大表面积,但催化剂外形不规整,可能呈无定型状态,导致催化活性较低;当浓度为0.625mol/L时,比表面积虽然变小,但颗粒均匀,外形规整近似呈球形,催化活性变大;当溶液浓度再高时,颗粒直径进一步增大,外形规整,比表面积进一步减小,催化活性降低。因此,规整外形和较大比表面积是其具有较好催化活性的前提。
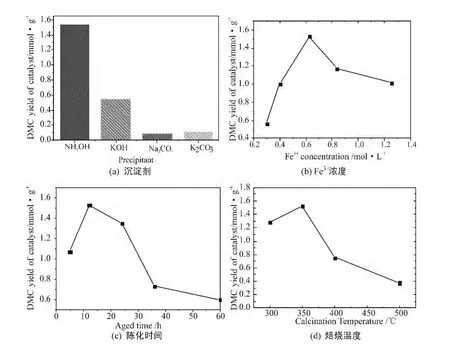
图7 催化剂制备条件对DMC收率的影响
2.2.3 陈化时间对DMC收率的影响
陈化过程是小晶粒溶解和大晶粒的长大过程,小晶粒的溶解再结晶使形成一次晶核中包裹的杂质释放,从而纯化晶体,因此陈化时间的长短直接影响催化剂的颗粒直径和杂质含量,从而影响催化剂的最终活性。以浓度为0.625mol/L的氨水为沉淀剂,水浴温度70℃,焙烧温度350℃时,考察不同陈化时间对DMC收率的影响,结果如图7(c)所示。陈化时间对DMC收率的影响也存在一峰值,即陈化时间为12h时,DMC的收率达到最大值。结合BET比表面积和SEM结果分析其原因为陈化时间较短时,颗粒不均匀,比表面积小,从而导致催化活性不高;陈化时间过长时,催化剂比表面积减小,催化活性下降。在最佳陈化时间即12h左右时,颗粒均匀,比表面积大,具有最高的催化活性。
2.2.4 焙烧温度对DMC收率的影响
从催化剂的表征结果可见,焙烧温度不仅影响颗粒大小,而且对催化剂的组成、形貌和晶型都有直接影响,从而对催化剂的催化活性产生影响。图7(d)为不同焙烧温度对催化剂催化合成DMC收率的影响结果。由图可知,随着焙烧温度的升高,DMC收率呈现先增大后减小的趋势,焙烧温度在350℃左右时,收率最高。结合IR、TG/DTA、N2吸附/脱附和XRD结果分析其原因为,由于氢氧化铁脱水没有固定温度,不同温度段所得催化剂的组成不同。当焙烧温度较低时,催化剂并非单组分Fe2O3,而可能是Fe2O3·xH2O,催化剂虽然比表面积大,但晶型不完整,所以催化活性较低;随着焙烧温度的提高,Fe2O3晶型趋于完整,催化活性提高,但催化剂BET比表面积下降而影响催化活性的提高。因此,在焙烧温度350℃左右出现最高催化活性,继续提高焙烧温度,催化剂的比表面积下降较快,催化活性降低。因此,完整晶型的Fe2O3是催化活性的主要因素。
2.3 脱水剂的加入对DMC收率的影响
完整晶型的Fe2O3在CO2与甲醇合成DMC的反应中表现出几乎接近100%的选择性,没有检测到副产物,但DMC收率较低,其主要原因是热力学平衡的限制[16]。如果能将生成的水移走从而打破平衡,即可获得较高的DMC收率。本部分采用文献中提到的乙腈[17-18]和N,N-二环己基碳二亚胺(DCC)[19]作为脱水剂,考察不同脱水剂及其加入量对DMC收率的影响,结果见表1。添加乙腈并没有使DMC的收率有很大提高,尤其是乙腈量少时,DMC收率反而略有下降。但添加DCC可使DMC收率明显增加,且随着DCC加入量的增加,DMC收率呈直线增加。分析其原因为在Fe2O3作用下,乙腈的水解速度较慢,甚至会降低其水解速度,但DCC由于其N=C=N中的碳具有较强的亲电性,很容易与水或甲醇中的O结合,而其所连结的N具有孤对电子,易形成氢键,因此很容易发生水解,水解的结果是快速消化掉产生的水,从而提高DMC收率。
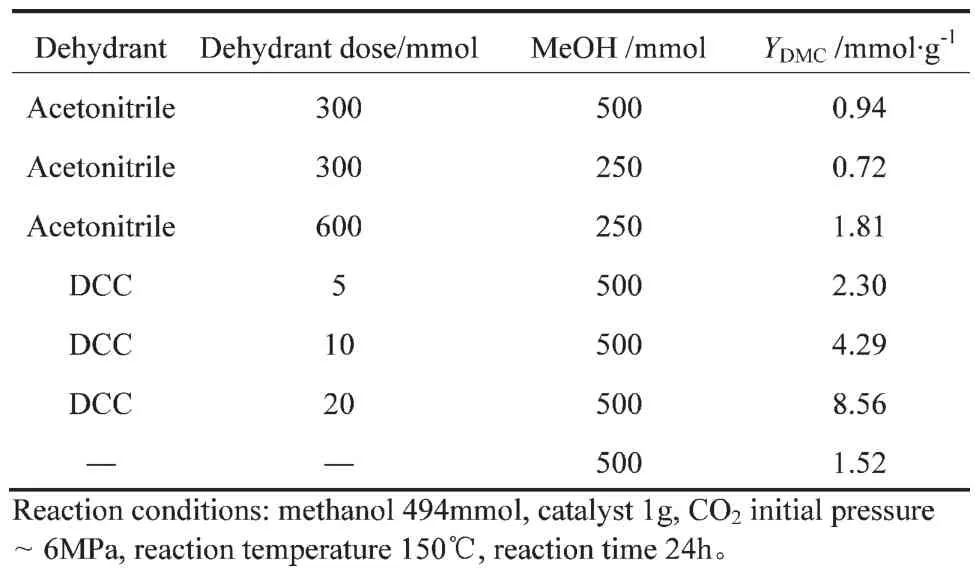
表1 脱水剂对Fe2O3催化剂的DMC收率的影响
2.4 催化剂的重复利用性能
对Fe2O3催化剂的重复利用性能进行了考察。将反应后的催化剂过滤,洗涤,在100℃干燥4h后作为催化剂继续进行活性实验,结果见图8。催化剂经过3次循环利用,活性变化不大,第4次后催化剂活性有所下降,但仍然具有较高的催化活性。
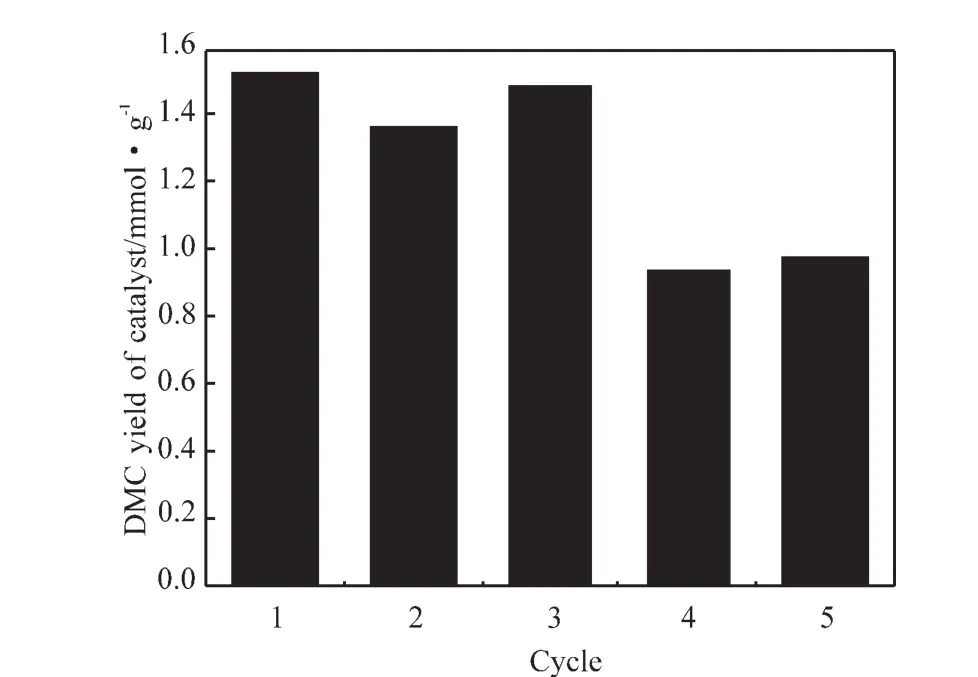
图8 催化剂循环次数对DMC收率的影响
通过对不同循环次数的催化剂进行XRD比较分析可知(见图9),催化剂反应前后的特征衍射峰没有变化,表现为典型的α-Fe2O3晶型,只是衍射峰强度变强,峰宽变窄,说明反应前后催化剂的晶型没有发生变化,晶粒变大。从图10(d)的SEM扫描图与反应前的比较可以看出,催化剂的颗粒略有变大,但形貌没有什么明显变化。从图10(a)~(c)TEM图可知,新鲜催化剂颗粒粘附性较大,颗粒堆积较密集,颗粒直径约在20nm~30nm之间,电子衍射环不明显,呈单晶的α-Fe2O3的衍射花纹;反应1次后,催化剂颗粒较分散,衍射环比新鲜催化剂明显,呈现类似多晶的衍射花纹,颗粒外形棱角较分明;反应3次后,催化剂颗粒较均匀,外形为花生壳形,电子衍射环完整,为典型的多晶衍射。由以上分析可知,完整晶型的α-Fe2O3晶型表现出较好的催化活性,循环次数不同晶型并没有改变,只是颗粒直径略有变化,从而使催化活性略有下降,所以催化剂可以重复利用。
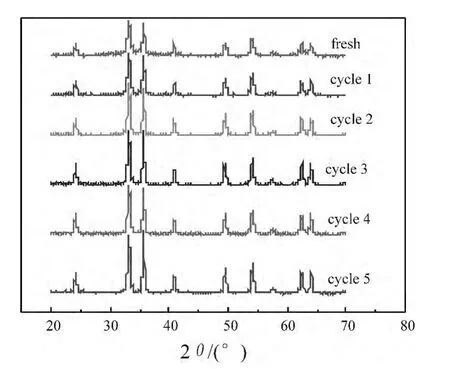
图9 反应前后催化剂的XRD谱图

图10 催化剂不同循环次数的TEM、电子衍射图谱和SEM
3 结论
氧化铁制备条件不同,所得形貌、晶型和比表面积亦不同,通过对沉淀剂、溶液浓度、陈化时间和焙烧温度的考察,以用于甲醇与CO2合成DMC反应中DMC的收率为评价指标,得到以氨水为沉淀剂、溶液中Fe3+浓度0.625mol/L,陈化时间12h、焙烧温度350℃时,所得催化剂活性最高,其DMC收率可达1.52mmol/g。通过SEM、XRD和N2吸附脱附分析发现,催化剂颗粒均匀,粒径在20nm~30nm,具有完整晶型和较大比表面积。添加脱水剂DCC可以使DMC收率明显提高,且随着DCC加入量的增加,DMC的收率呈直线上升趋势,当DCC加入量为20mmol时,DMC收率提高了近5倍。催化剂循环利用5次,催化活性降低很少。
[1]Castro A J R,Soares J M,Filho J M,et al.Oxidative dehydrogenation of ethylbenzene with CO2for styrene production over porous iron-based catalysts[J].Fuel,2013,108:740-748.
[2]Liu,Y,Sun D.Effect of CeO2doping on catalytic activity of Fe2O3/gamma-Al2O3catalyst for catalytic wet peroxide oxidation of azo dyes[J].J Hazard Mater,2007,143(1-2):448-454.
[3]Ren X,Chang L,Li F,et al.Study of intrinsic sulfidation behavior of Fe2O3for high temperature H2S removal[J].Fuel,2010,89(4):883-887.
[4]Wu G,Tan X,Li G,et al.Effect of preparation method on the physical and catalytic property of nanocrystalline Fe2O3[J].J Alloy Compd,2010,504(2):371-376.
[5]Wheeler D A,Wang G,Ling Y,et al.Nanostructured hematite: synthesis, characterization, charge carrier dynamics,and photoelectrochemical properties[J].Energ Environ Sci,2012,5(5):6682-6702.
[6]肖雪,孔龙江,路嫔.尿素醇解法合成DMC中纳米氧化铁催化剂制备工艺 [J].黑龙江工程学院学报,2010,2(1):46-49.
[7]Zhou Y,Wang S,Xiao M,et al.Novel Cu-Fe bimetal catalyst for the formation of dimethyl carbonate from carbon dioxide and methanol[J].RSC Adv,2012,2(17):6831-6837.
[8]Lee H J,Joe W,Song I K.Direct synthesis of dimethyl carbonate from methanoland carbon dioxide over transition metal oxide/Ce0.6Zr0.4O2catalysts:Effect of acidity and basicity of the catalysts[J].Korean J Chem Eng,2012,29(3):317-322.
[9]Watanabe M,Kosuge O,Inomata H.Study of direct synthesis of dimethyl carbonate from supercritical carbon dioxide and methanol using metal oxide catalyst[J].Chorinkai Saishin Gijutsu,2003,7:60-65.
[10]巩志坚,田原宇,李文华,等.铁氧化物的热特性研究[J].洁净煤技术,2006,(3):95-97
[11]Zhan S,Chen D,Jiao X,et al.Facile fabrication of long α-Fe2O3, α-Fe and γ-Fe2O3hollow fibers using sol– gel combined co-electrospinning technology[J].J Colloid Interf Sci,2007,308(1):265–270
[12]Wu G,Tan X,Li G,et al.Effect of preparation method on the physical and catalytic property of nanocrystalline Fe2O3[J].J Alloy Compd,2010,504(2):371-376
[13]张怡.氧化铁纳米结构的水热/溶剂热合成及其催化性能的表征[D].浙江:浙江大学硕士学位论文,2008:46.
[14]Liu X M,Fu S Y,Xiao H S,et al.Preparation and characterization of shuttle-like α-Fe2O3nanoparticles by supermolecular template[J].J Solid State Chem,2005,178(9):2798-2803.
[15]高瞻,董晟全,梁艳峰,等.超细纳米氧化铁粉体的制备与表征[J].纳米科技,2011,8(6):53-56.
[16]Cai Q,Lu B,Guo L,et al.Studies on synthesis of dimethyl carbonate from methanol and carbon dioxide[J].Catal Commun,2009,10(5):605-609
[17]Honda M,Suzuki A,Noorjahan B,et al.Low pressure CO2to dimethyl carbonate by the reaction with methanol promoted by acetonitrile hydration [J].Chem Commun,2009,(30):4596-4598.
[18]Honda M,Kuno S,Begum N,et al.Catalytic synthesis of dialkyl carbonate from low pressure CO2and alcohols combined with acetonitrile hydration catalyzed by CeO2[J].Appl Catal A,2010,384(1-2):165-170
[19]lsaecs N S,O'Sullivan B,Verhaelen C.High pressure routes to dimethyl carbonate from supercritical carbon dioxide[J].tetrahedron,1999,55(40):11949-11956.
猜你喜欢
青岛科技大学学报(自然科学版)(2023年6期)2023-11-25 17:17:56
Acta Mathematica Scientia(English Series)(2021年1期)2021-04-08 12:52:22
陶瓷学报(2020年2期)2020-10-27 02:16:14
中国茶叶加工(2020年3期)2020-10-21 08:06:48
河北工业大学学报(2019年3期)2019-09-10 10:46:34
中国塑料(2015年6期)2015-11-13 03:02:34
中国塑料(2015年8期)2015-10-14 01:10:48
科技创新导报(2014年20期)2014-11-10 05:35:05
无机盐工业(2014年6期)2014-06-11 01:58:30
中国烟草学报(2012年4期)2012-04-09 07:11:48
