
腓骨肌萎缩症4型遗传学研究进展
2015-10-13 08:25许烨张嘉莹杨博宇何志宏张慕晨于珍顾鸣敏
遗传 2015年6期
许烨,张嘉莹,杨博宇,何志宏,张慕晨,于珍,顾鸣敏
1.上海交通大学医学院,上海 200025;
2.上海交通大学医学院医学遗传学教研室,上海 200025
腓骨肌萎缩症4型遗传学研究进展
许烨1,张嘉莹1,杨博宇1,何志宏1,张慕晨1,于珍2,顾鸣敏2
1.上海交通大学医学院,上海 200025;
2.上海交通大学医学院医学遗传学教研室,上海 200025
腓骨肌萎缩症也称夏科-马利-杜斯氏病(Charcot-Marie-Tooth disease,CMT),是人类最常见的遗传性周围神经病之一,其遗传方式以常染色体显性遗传为主,也有部分呈常染色体隐性遗传或X连锁显性或隐性遗传。根据临床表型将CMT分为脱髓鞘型(CMT1)、轴突型(CMT2)和中间型(DI-CMT)。常染色体隐性遗传的CMT1(AR-CMT1,也称CMT4型)临床表现除了CMT常见的四肢远端进行性肌无力和萎缩,以及高足弓和爪形手外,常起病早,进展迅速,并有不同程度的感觉障碍和脊柱畸形(以脊柱侧凸为主)。近年来的研究显示,CMT4有11种亚型,其中有些亚型的致病机制较明确,有些亚型存在建立者突变,有些亚型还局限在临床描述和突变检出上。文章综述了CMT4的最新研究进展,包括各亚型的临床表现、致病机制和小鼠模型等。
腓骨肌萎缩症;CMT4型;致病机制;小鼠模型
1 CMT4的临床特点及其分型
CMT4在地中海沿岸的国家较为常见,且存在表型异质性。临床表现除了CMT常见的四肢远端进行性肌无力和萎缩,以及高足弓和爪形手外,还有一些特征性表现,如多数患者幼年起病,进展迅速,症状严重,伴有不同程度的感觉障碍和脊柱畸形(以脊柱侧凸为主),导致明显的远端肢体畸形,最终丧失行走能力[4]。少数患者也出现横隔瘫痪、青光眼、失聪、无精症等表现。神经电生理检查显示上肢MNCV低于正常,神经活检显示显著的髓鞘异常,以节段性脱髓鞘、施万细胞增生、呈“洋葱头”样改变为特征。此外,CMT4呈AR遗传,在近亲婚配家系的后代中患病率较高。CMT4也存在遗传异质性。根据受累基因(包括GDAP1、MTMR2、MTMR13、MTMR5、SH3TC2、NDRG2、EGR2、PRX、HK1、
FDG4和FIG4)发现的先后顺序,已将其分为11种亚型,分别是CMT4A、4B1、4B2、4B3、4C、4D、4E、4F、4G、4H和4J[2]。CMT4各亚型的致病基因及其定位、发病年龄及临床特点见表1。值得一提的是,根据临床表型无法区分CMT4各亚型,确诊依赖于基因突变分析结果。
2 CMT4各亚型致病机制的研究进展
2.1 CMT4A
CMT4A约占所有CMT4的25%,是最常见的一种亚型。Baxter等[5]对4个突尼斯家系进行了研究,将该病的致病基因——神经节苷脂诱导分化相关蛋白 1(Ganglioside-induced differentiation-associated protein-1,GDAP1)定位于8q21.3上。除了CMT4A,GDAP1突变还可见于其他CMT亚型,包括引起声音嘶哑的AR-CMT2K(轴突型隐性腓骨肌萎缩症)、CMTRIA(中间型隐性腓骨肌萎缩症)和罕见的AD-CMT2K(显性腓骨肌萎缩症亚型)。
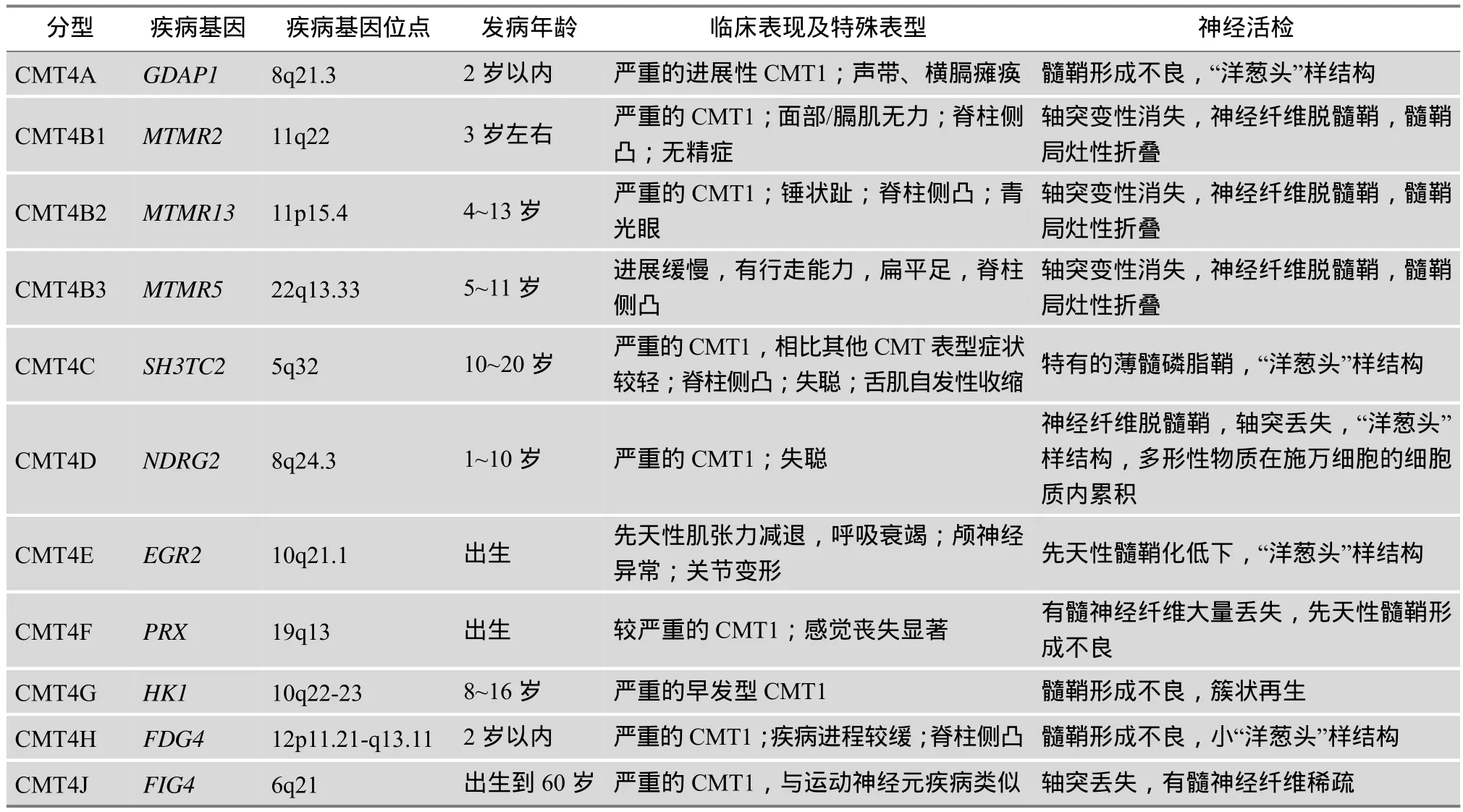
表1 CMT4各亚型的致病基因及临床特点
研究发现,GDAP1是表达在周围神经和中枢神经系统多个区域的施万细胞(Schwann cell)和神经元线粒体外膜的膜蛋白,这种膜蛋白对调控有髓外周神经中的线粒体分裂和融合的动态变化有着重要的作用[6],而维持线粒体的动态平衡是有髓神经发挥正常功能的重要因素[7]。Niemannet等[8]研究发现隐性遗传的GDAP1突变减弱了线粒体的分裂能力。与之相反,显性遗传的GDAP1突变则减弱了线粒体的融合能力。线粒体的动态变化异常将干扰线粒体的运输及生物合成,导致线粒体DNA的损失、线粒体膜电位的下降和活性氧族(Reactive oxygen species, ROS)水平的增加。最近研究表明,GDAP1与细胞内抗氧化剂谷胱甘肽含量及线粒体活动控制相关,提示CMT4A的致病机制可能与氧化应激相关[9],但是具体分子机制尚未明确。
研究还发现,GDAP1基因突变主要集中在地中海沿岸国家,其中北非国家最常见的突变(p.Ser194*)在西班牙也有发现,故将该突变称为建立者突变(Founder mutation)[5]。此外,p.Gln163*突变是北美西班牙裔人群中最常见的突变。
GDAP1基因突变所导致的神经病变较为复杂,兼有脱髓鞘和轴突变性,由此导致该病临床表现的高度异质性。这种表型异质性不仅体现在相同突变位点患者的起病年龄和功能障碍的不同[5],同时也体现在家系成员之间表型的差异。由于该基因的大多数突变仅见于少数无血缘关系的家系或患者,目前尚难以确定GDAP1基因型与表型之间的相关性。但是如果患者起病早,出现严重的轴突病变或脱髓鞘,加上严重的足部畸形常提示可能为GDAP1基因突变所致。如果患者来自摩洛哥或西班牙,应检测是否存在p.Ser194*和p.Gln163*突变[5]。
2.2 CMT4B1
Tazir等[10]曾报道患 CMT4B1的家族成员在MTMR2基因存在1个c.331dupA(p.Arg111LysfsX24)的纯合突变。神经病理学研究显示,CMT4B1存在髓磷脂外折的表现。
为了阐明该基因突变导致CMT4B1的分子机制,Bolino等[11]构建了一种新的小鼠模型:敲除Mtmr2的4号外显子,导致位于1号或3号外显子的ATG在翻译时发生移码突变,由此产生一段没有功能的短肽;行为学及神经生理学分析显示,Mtmr2敲除小鼠存在周围神经疾病。此外,Bolino等[11]从大鼠Mtmr2全长cDNA中找到了Dlg1基因及 SAP97基因与CMT4B1相关。进一步的转染和免疫共沉淀实验表明MTMR2与Dlg1/SAP97共定位,这提示在施万细胞中MTMR2与Dlg1/SAP97有着生理功能上的联系。Bolino等[11]基于如下3点提示MTMR2缺陷时施万细胞可发生结构异常:(1)髓磷脂可外折,并在郎飞结旁区域(Paranode region)占有主导地位;(2)MTMR2与Dlg1/SAP97相互作用,其中后者为在施万细胞中发现的小分子但不存在于轴突中,其在野生型的郎飞结旁和微绒毛中表达丰富,但在MTMR2阴性的纤维中不表达;(3)去除了施万细胞的Mtmr2的表现型与Mtmr2敲除的小鼠相一致。
2.3 CMT4B2
CMT4B2型由MTMR13突变所致,该基因编码SBF2(SET binding factor 2),也称为肌微管素相关蛋白13(MTMR13)。
研究显示,MTMR2和MTMR13的缺失均可导致髓磷脂折叠失控,其机制尚未明确。但已知同型二聚体MTMR2作为一种磷酸肌醇维生素D3-磷酸酶(PIs),以磷脂酰肌醇3-磷酸(PtdIns3P)和磷脂酰肌醇3,5-二磷酸(PtdIns[3,5]P2)为底物,后两者的作用是在溶酶体途径中调节膜流量。MTMR13无催化活性,但可以与MTMR2形成四聚体复合物,使MTMR2的酶促活性显著增强[12]。而PIs作为膜栓系的信号分子,调节细胞分裂、细胞生长和存活、细胞内的膜运输、肌动蛋白活性及其信号传导等过程。SBF2功能的缺失将导致MTMR2的功能丧失,引起磷脂酰肌醇3-磷酸和磷脂酰肌醇3,5-二磷酸的堆积,使膜运输等途径失调,影响施万细胞之间的相互作用[13]。
现有两种CMT4B2的小鼠模型,都利用基因捕获技术以产生突变的Mtmr13。Tersar等[14]在Mtmr13的14号内含子处插入一段序列,而Robinson等[15]则在Mtmr13的16、17号内含子之间缺失了一段3564 bp的序列。Mtmr13–/–小鼠再现了人类CMT4B2疾病的几个关键表型,即MNCV减慢、复合肌肉动作电位幅度减小,以及髓磷脂外翻和内折,这些表现随着年龄增长而加重。神经传导速度的改变较轻微,髓磷脂层的厚度和轴突直径未见明显改变。28个月的Mtmr13–/–小鼠外周神经组织中可以观察到明显的轴突变性,髓鞘化低下和节段性脱髓鞘-髓鞘再生,所有这些都是人类CMT4B2的病理特征[16]。Robinson等[15]还发现在Mtmr13–/–小鼠的坐骨神经中Mtmr2蛋白水平降低了50%,提示MTMR2和MTMR13之间存在密切的调控关系。
2.4 CMT4B3
CMT4B3亚型是由Nakhro等[17]通过对一个韩国家系的全外显子组测序中确定并命名的。在该家系中,3位患者的发病年龄在5~11岁之间,表现出腿部远端肌无力,并呈跨越步态,伴有爪状手及脊柱侧弯等现象。但是该病进展缓慢,不影响患者行走能力。通过外显子组测序发现其潜在的病因为SET结合因子1(SBF1)基因(也称为MTMR5)上的杂合错义突变[c.A1249G(p.M417V)]+[c.A4768G(p.T1590A)]。
SBF1/MTMR5在结构上与SBF2/MTMR2相似。同时SBF1也和其他肌管相关蛋白具有相似的功能。目前的假说是SBF1与内涵体的运输有关。SBF1与MTMR2通过卷曲螺旋结构域相互作用,影响其亚细胞定位,并增加MTMR2的酶活性,从而在运动神经细胞中发挥作用。
2.5 CMT4C
CMT4C是由SH3TC2/KIAA1985基因突变所致,基因产物是一种含有多个SH3(Src homology domain)和TPR(Tetratrieopeptide repeat)结构域的蛋白质,可能是一种多蛋白复合物的成分之一。SH3结构域能识别富含脯氨酸和疏水残基的特异序列的蛋白质并与之结合。TPR基序是一个含有34个氨基酸的重复序列,通常串联排列。两种结构域形成特殊的空间结构,以介导蛋白质的相互作用,并在一些重要的蛋白复合物中起作用。迄今为止,在地中海沿岸国家及欧洲地区已有超过20个突变位点被报道,其中最频发的突变为Arg1109*和Arg954*。Arg954*突变常发生在法裔加拿大人和英国人的家庭中[18],而Arg1109*突变常发生在西班牙裔的吉普赛人中[19]。特别是Arg954*突变常表现为严重的脊柱侧凸,故也将脊柱病变作为CMT4C的标志性特征之一。
1996年,Leguern等[20]发现CMT4C的突变区域在5q23-33。该型的发病时间在1~12岁之间,更大的范围是在2~50岁之间[21]。疾病呈缓慢进行性发展,相比其他亚型来说,CMT4C的症状较轻。日本一名女性患者从高中开始出现脊柱侧凸的表现,20岁时手部畸形,但直至69岁才出现足部畸形[22]。脊柱畸形是这一亚型的特征性表现,约80%的患者都会发展成严重的脊柱侧凸而需要手术治疗。腓肠神经的病理学显示特有的薄髓磷脂鞘和施万细胞大量增生并出现“洋葱头”样改变[23]。还有一些患者有听觉迟钝和轻度面瘫的症状[24]。
Yger等[21]发现SH3TC2基因参与脊髓的发育,这或许可以解释CMT4C患者脊椎侧凸的原因。有关SH3TC2编码的蛋白质功能和致病机制的研究尚在进行中。多项研究均发现SH3TC2定位于细胞内胞吞途径相关蛋白,同时也表达于细胞膜上[25,26]。2010年,Stendel等[27]发现SH3TC2是Rab11(一种小GTP酶)的新效应分子。SH3TC2通过与Rab11相互作用定位于细胞内再循环的内体。同时,野生型SH3TC2影响转铁蛋白受体的动态变化,而带有SH3TC2突变的细胞无法与Rab11结合,失去了定位及转铁蛋白受体动力学的能力。这提示SH3TC2和Rab11结合引起的效应可能就是CMT4C患者致病的分子机制。但是,目前尚未发现SH3TC2与Rab11结合的肽段。
在Sh3tc2基因敲除小鼠中发现Sh3tc2缺失使得与髓鞘生成相关基因的表达下调,包括髓鞘碱性蛋白(MBP)、髓鞘蛋白零(MPZ)和胆固醇合成的基因等[18]。同时研究也发现敲除Sh3tc2基因的小鼠郎飞结加长,有利于人们更方便地鉴定出CMT4C[18]。
2.6 CMT4D
Kalaydjieva等[28]最先报道CMT4D与位于8q24.3的NDRG1基因突变存在相关性,随后他又发现了该基因的一个无义突变g.631C>T(p.Arg148*)[29],该突变也被认为是CMT4D的建立者突变。
研究发现,该基因编码的蛋白质NDRG1可以使Nmyc的表达下调,是参与应激反应、激素反应、细胞生长和分化的胞质蛋白,是一种细胞内多泡体形成和低密度脂蛋白受体(Low density lipoprotein receptors,LDLR)胞内转运的调节器。NDRG1的突变可能会影响血脂的处理和髓鞘细胞的分化。在NDRG1被沉默的上皮细胞中,由于质膜上LDLR丰度降低使上皮细胞摄取的低密度脂蛋白(Low density lipoprotein,LDL)减少,结果导致LDLR在含大量腔内囊泡和螯合酰胺的EEA1阳性核内体中的积累。与此同时,LDLR泛素化增加且降解减少,ESCRT(转运必需内吞体分选复合物)蛋白被下调,耗竭IDOL(低密度脂蛋白的诱导降解物),从而改变质膜上LDLR的含量和LDL的摄取量[29]。
在Pietiäinen等[30]构建的小鼠模型中,NDRG1沉默小鼠的少突胶质细胞中LDL摄取降低,少突细胞分化因子OLIG2水平同时下调,这两种表型都由沉默NDRG1引起,提示NDRG1可通过LDLR家族成员的配体的吸收来控制少突胶质细胞的分化。
2.7 CMT4E
CMT4E是由EGR2基因的纯合突变所导致的一种罕见的先天性神经病变,最早由Warner等[31]于1998年报道。EGR2基因编码早期生长反应蛋白2(Early growth response protein 2,EGR-2)。EGR2是一个转录因子,包含3个串联的Cys2His2型锌指结构,可以在外周神经系统的髓鞘蛋白,如髓鞘蛋白零(Myelin Protein Zero)、连接蛋白 32(CX 32)和periaxin蛋白[32]的调节中发挥作用,因此EGR2有控制外周神经系统髓鞘形成的作用[33]。该基因的多态性与CMT4E、CMT1及进行性肥大性间质性神经病(DSS)相关,其突变造成的功能缺失引起的髓鞘发育不良为CMT4E的主要致病因素。Decker等[34]的研究显示,EGR2基因突变的成年小鼠施万细胞产生严重脱髓鞘,包括施万细胞的去分化和增殖增加。因此证明EGR2基因对髓鞘的形成及其功能状态的维持是至关重要的。
2.8 CMT4F
导致CMT4F的致病基因PRX定位于19q13.1-19q13.3。PRX编码能保护周围神经髓磷脂的蛋白质,该蛋白有两个PDZ结构域,有两个转录变异可以编码不同的蛋白异构体,并且对施万细胞的作用靶点不尽相同。CMT4F在多个种族中均被发现,例如黎巴嫩什叶派穆斯林,北美洲的西班牙后裔、北欧人、越南人[35]。截止目前,已报道有16个PRX基因的无义或移码突变可导致CMT4F[36]。
为了阐明PRX突变导致CMT4F的分子机制,Williams等[37]构建了Prx基因敲除小鼠模型。通过对该小鼠模型以及PRX的蛋白-蛋白反应的研究,发现PRX在稳定髓磷脂鞘中起到不可或缺的作用。人为对6周大的Prx基因敲除小鼠的坐骨神经进行损伤,6周后,其损伤侧轴突的数量可恢复正常,但其直径比对侧轴突小。与正常小鼠相比,Prx敲除小鼠不仅有更多小直径的轴突,而且在神经再生时继续发生。
2.9CMT4G
CMT4G(HMSN-Russe)是西班牙吉普赛人CMT患者中的第二大亚型,仅次于CMT4C[38]。CMT4G由位于10q22-23的HK1基因突变所致。已知该基因产物HK1蛋白通过其孔蛋白结合域与线粒体膜结合,催化葡萄糖发生磷酸化生成G-6-P,调节细胞能量代谢,在大脑、睾丸、红细胞中含量丰富。
2009年,Hantke等[39]发现了两个完全连锁不平衡的序列变异,即交替外显子AltT2的G>C突变和其相邻内含子的G>A突变。通过对16个物种的序列分析发现,Alt2突变位点处的碱基G保持不变,具有极高的保守性。这强烈提示该突变为致病突变,称为HK1 g.9712G>C。若HK1的编码序列发生突变,会导致葡萄糖磷酸酶的缺陷及溶血性贫血。然而,研究也发现HMSNR患者施万细胞中的酶含量与对照组相比并未下降,即患者体内的HK1蛋白在细胞能量代谢中的催化调节作用并未受抑。目前,HK1 AltT2突变的致病机制仍未知。
2.10 CMT4H
CMT4H由FGD4基因突变所致,其编码的FRABIN蛋白是一种鸟嘌呤核苷酸交换因子,对Cdc42分子有特异性,能激活Rho GTP酶。该酶在真核生物应答外界或内部信号时,使GDP转化为有活性的GTP形式,发挥信号转导作用;同时,通过影响施万细胞的细胞迁移、形态发生、极化、分裂、囊泡-膜运输,影响周围神经系统的髓鞘形成。FRABIN蛋白由766个氨基酸构成,包含5个结构域,即N¢端的f肌动蛋白结合域、一个DH(Dbl Homology)结构域、两个PH(Pleckstrin Homology)结合域以及一个富含半胱氨酸的FYVE结构域。DH结构域催化GDP转化成GTP,而PH和FYVE结构域主要参与磷酸肌醇不同形式的转化。
2007年,Delague等[40]在一个黎巴嫩家系中发现的突变位点为c.893T>G(p.Met298Arg),而在另一个阿尔及利亚家族中发现的突变位点为c.893T>C (p.Met298Thr)。这两个突变位点都位于DH结构域,但p.Met298Thr突变可能改变结构域的组成,从而阻止FRABIN与Cdc42的结合。而p.Met298Arg属于剪接位点突变,其编码产物是一种截短蛋白。在Horn等[41]构建的Frabin突变小鼠模型中,小鼠施万细胞中FRABIN/FGD4未能表达,且在60周时出现神经电生理缺陷,表现为神经传导速度下降,出现F波等。实验证明该蛋白对神经发育和髓鞘维持是不可或缺的。
2.11 CMT4J
大部分无血缘关系的CMT4J患者的基因型是由一个独特的无效等位基因FIG4和共有的来自祖先的错义等位基因FIG4-I41T组合而成的复合杂合子[42]。FIG4基因位于6q21上,编码一种定位于细胞膜上的聚磷酸肌醇磷酸酶FIG4,参与磷酸肌醇含量和囊泡运输的调节。此外,FIG4杂合子突变也能导致肌萎缩性侧索硬化症[43]。
Chow等[44]在小鼠的杂交品系中发现了一种有严重震颤、步态异常的突变小鼠,命名为“苍白震颤”小鼠(plt)。进一步分析发现plt小鼠是由ETn2β(转座子2β)插入到Fig4第18号外显子而产生的无效突变体。Plt小鼠出现了类似CMT的临床表现和病理特征:小鼠步态异常,外周神经受累,坐骨神经传导速度减慢和复合肌肉动作电位幅度减小,还出现了大直径的有髓轴突数量减少的现象。
对plt小鼠成纤维细胞磷酸肌醇的分析表明其PtdIns(3,5)P2水平为正常的1/3。40%的plt小鼠成纤维细胞充满了增大的晚期内体囊泡。进一步研究发现FIG4编码蛋白能够特异性地去除从PI(3,5)P2上肌醇相连的5-磷酸基团的一种磷酸酶。FIG4通过与另2种蛋白质(PIKfyve和VAC14)的相互作用,形成一种蛋白质复合物来调节PI(3,5)P2合成。FIG4突变会降低FIG4与另2种蛋白质的亲和力,从而影响PI(3,5)P2的含量。而PI(3,5)P2是一种定位于核内体/溶酶体途径的小泡胞质面的磷脂,参与细胞内囊泡的运输和融合的信号转导。研究还表明I41T突变削弱了FIG4与脚手架蛋白VAC14的相互作用[45]。在VAC14缺失的小鼠中FIG4蛋白的减少可以证实这一相互作用。对患者成纤维细胞的分析表明,突变体I41T蛋白质处于相对较低的水平,而在加入蛋白酶体抑制剂MG-132培养的细胞中,FIG4-I41T蛋白的丰度有所增加。该研究提示,FIG4-I41T是亚效等位基因,其编码的蛋白质在体内无法稳定存在。
3 结 语
CMT4曾被认为是极其罕见且种类很少。然而最近的临床和分子遗传学研究已确定了10余种新的基因突变及其相应的蛋白质,为研究人员更全面了解这类疾病的病理生理学特征奠定了基础。值得注意的是,80%以上的AD-CMT的分子诊断仅限于发病率较高的PMP22、MPZ、GJB1和MFN2,而对于发病率较低的CMT4型的分子诊断更具挑战性[10]。目前的策略是先通过流行病学、临床及病理检查作筛选,然后进行分子诊断,例如出现声带麻痹、面部及延髓麻痹、早期青光眼、严重的脊柱侧弯同时伴有早期发病或严重病变往往提示与GDAP1、MTMR2、MTMR13、SH3TC2基因突变所致的CMT4型有关[46]。此外,髓鞘折叠提示存在MTMR2、MTMR13或PRX突变,而基底膜“洋葱头”样改变或出现异常的郎飞结结构提示SH3TC2基因发生了突变。
有关机制研究还需要补充的是,涉及CMT4致病机制的蛋白质的生物学功能是多样的,如参与聚磷酸肌醇的信号转导和膜运输蛋白包括MTMR2、MTMR13、FIG4和SBF1,控制线粒体分裂的蛋白有GDAP1,髓鞘结构蛋白有periaxin(L-PRX)和EGR2,参与生长停滞和细胞分化蛋白质有NDRG1,以及细胞内吞作用动力学和运输相关蛋白质有SH3TC2、NDRG1。迄今,只有少数CMT4亚型的致病机制已被揭示,而其他基因的致病机制尚有待进一步研究,任重而道远。
[1]Lupski JR,Reid JG,Gonzaga-Jauregui C,Rio Deiros D, Chen DC,Nazareth L,Bainbridge M,Dinh H,Jing C, Wheeler DA,McGuire AL,Zhang F,Stankiewicz P, Halperin JJ,Yang C,Gehman C,Guo D,Irikat RK,Tom W, Fantin NJ,MuznyDM,GibbsRA.Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy.N Engl J Med,2010,362(13):1181–1191.
[2]Rossor AM,Polke JM,Houlden H,Reilly MM.Clinical implications of genetic advances in Charcot-Marie-Tooth disease.Nat Rev Neurol,2013,9(10):562–571.
[3]Xu WY,Gu MM,Sun LH,Guo WT,Zhu HB,Ma JF,Yuan WT,Kuang Y,Ji BJ,Wu XL,Chen Y,Zhang HX,Sun FT, Huang W,Huang L,Chen SD,Wang ZG.A nonsense mutation in DHTKD1 causes Charcot-Marie-Tooth disease type 2 in a large Chinese pedigree.Am J Hum Genet,2012, 91(6):1088–1094.
[4]Claeys KG,Lammens M,Senderek J,Weis J.Autosomal recessive demyelinating or axonal Charcot-Marie-Tooth neuropathy.In:Vallat JM,Director JW.Peripheral nerve disorders:pathology and genetics.UK:The International Society of Neuropathology,2014:85–101.
[5]Baxter RV,Ben Othmane K,Rochelle JM,Stajich JE, Hulette C,Dew-Knight S,Hentati F,Ben Hamida M,Bel S, Stenger JE,Gilbert JR,Pericak-Vance MA,Vance JM. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat Genet,2002,30(1):21–22.
[6]Niemann A,Ruegg M,La Padula V,Schenone A,Suter U. Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network:new implications for Charcot-Marie-Tooth disease.J Cell Biol,2005,170(7): 1067–1078.
[7]Pedrola L,Espert A,Wu XY,Claramunt R,Shy ME,Palau F.GDAP1,the protein causing Charcot-Marie-Tooth disease type 4A,is expressed in neurons and is associated with mitochondria.Hum Mol Genet,2005,14(8):1087–1094.
[8]Niemann A,Huber N,Wagner KM,Somandin C,Horn M, Lebrun-Julien F,Angst B,Pereira JA,Halfter H,Welzl H, Feltri ML,Young P,Wessig C,Toyka KV,Suter U.The Gdap1 knockout mouse mechanistically links redox control to Charcot-Marie-Tooth disease.Brain,2014,137(Pt 3): 668–682.
[9]Noack R,Frede S,Albrecht P,Henke N,Pfeiffer A,Knoll K,Dehmel T,Meyer Zu Hörste G,Stettner M,Kieseier BC, Summer H,Golz S,Kochanski A,Wiedau-Pazos M, Arnold S,Lewerenz J,Methner A.Charcot-Marie-Tooth disease CMT4A:GDAP1 increases cellular glutathione and the mitochondrial membrane potential.Hum Mol Genet,2012,21(1):150–162.
[10]Tazir M,Bellatache M,Nouioua S,Vallat JM.Autosomal recessive Charcot-Marie-Tooth disease:from genes to phenotypes.J Peripher Nerv Syst,2013,18(2):113–129.
[11]Bolino A,Bolis A,Previtali SC,Dina G,Bussini S,Dati G, Amadio S,Del Carro U,Mruk DD,Feltri ML,Cheng CY, Quattrini A,Wrabetz L.Disruption of Mtmr2 produces CMT4B1-like neuropathy with myelin outfolding and impaired spermatogenesis. J Cell Biol, 2004, 167(4):711–721.
[12]Berger P,Berger I,Schaffitzel C,Tersar K,Volkmer B, Suter U.Multi-level regulation of myotubularin-related protein-2 phosphatase activity by myotubularin-related protein-13/set-binding factor-2.Hum Mol Genet,2006, 15(4):569–579.
[13]Robinson FL, Dixon JE. The phosphoinositide-3-phosphatase MTMR2 associates with MTMR13,a membrane-associated pseudophosphatase also mutated in type 4B Charcot-Marie-Tooth disease.J Biol Chem,2005,280(36):31699–316707.
[14]Tersar K,Boentert M,Berger P,Bonneick S,Wessig C, Toyka KV,Young P,Suter U.Mtmr13/Sbf2-deficient mice: an animal model for CMT4B2.Hum Mol Genet,2007, 16(24):2991–3001.
[15]Robinson FL,Niesman IR,Beiswenger KK,Dixon JE. Loss of the inactive myotubularin-related phosphatase Mtmr13 leads to a Charcot-Marie-Tooth 4B2-like peripheral neuropathy in mice.Proc Natl Acad Sci USA,2008, 105(12):4916–4921.
[16]Ng AA,Logan AM,Schmidt EJ,Robinson FL.The CMT4B disease-causing phosphatases Mtmr2 and Mtmr13 localize to the Schwann cell cytoplasm and endomembrane compartments,where they depend upon each other to achieve wild-type levels of protein expression.Hum Mol Genet,2013,22(8):1493–1506.
[17]Nakhro K,Park JM,Hong YB,Park JH,Nam SH,Yoon BR,Yoo JH,Koo H,Jung SC,Kim HL,Kim JY,Choi KG, Choi BO,Chung KW.SET binding factor 1(SBF1) mutation causes Charcot-Marie-Tooth disease type 4B3. Neurology,2013,81(2):165–173.
[18]Arnaud E,Zenker J,de Preux Charles AS,Stendel C,Roos A,Médard JJ,Tricaud N,Kleine H,Luscher B,Weis J, SuterU,SenderekJ,ChrastR.SH3TC2/KIAA1985 protein is required for proper myelination and the integrity of the node of Ranvier in the peripheral nervous system. Proc Natl Acad Sci USA,2009,106(41):17528–17533.
[19]Colomer J,Gooding R,Angelicheva D,King RHM, Guillén-Navarro E,Parman Y,Nascimento A,Conill J, Kalaydjieva L.Clinical spectrum of CMT4C disease in patients homozygous for the p.Arg1109X mutation in SH3TC2.Neuromuscul Disord,2006,16(7):449–453.
[20]LeGuern E,Guilbot A,Kessali M,Ravisé N,Tassin J, Maisonobe T,Grid D,Brice A.Homozygosity mapping of an autosomal recessive form of demyelinating Charcot-Marie-Tooth disease to chromosome 5q23-q33. Hum Mol Genet,1996,5(10):1685–1688.
[21]Yger M,Stojkovic T,Tardieu S,Maisonobe T,Brice A, Echaniz-Laguna A,Alembik Y,Girard S,Cazeneuve C, LeGuern E,Dubourg O.Characteristics of clinical and electrophysiological pattern of Charcot-Marie-Tooth 4C.J Peripher Nerv Syst,2012,17(1):112–122.
[22]Iguchi M,Hashiguchi A,Ito E,Toda K,Urano M,Shimizu Y,Takeuchi C,Saito K,Takashima H,Uchiyama S. Charcot-marie-tooth disease type 4C in Japan:Report of a case.Muscle Nerve,2013,47(2):283–286.
[23]Houlden H,Laura M,Ginsberg L,Jungbluth H,Robb SA, Blake J,Robinson S,King RHM,Reilly MM.The phenotype of Charcot–Marie–Tooth disease type 4C due to SH3TC2 mutations and possible predisposition to an inflammatory neuropathy.NeuromusculDisord,2009, 19(4):264–269.
[24]Kalaydjieva L,Gresham D,Gooding R,Heather L,Baas F, de Jonge R,Blechschmidt K,Angelicheva D,Chandler D, Worsley P,Rosenthal A,King RHM,Thomas PK.N-myc downstream-regulated gene 1 is mutated in hereditary motor and sensory neuropathy-Lom.Am J Hum Genet, 2000,67(1):47–58.
[25]Roberts RC,Peden AA,Buss F,Bright NA,Latouche M, Reilly MM,Kendrick-Jones J,Luzio JP.Mistargeting of SH3TC2awayfrom therecycling endosomecauses Charcot-Marie-Tooth disease type 4C.Hum Mol Genet, 2010,19(6):1009–1018.
[26]Lupo V,Galindo MI,Martínez-Rubio D,Sevilla T,Vílchez JJ,Palau F,Espinós C.Missense mutations in the SH3TC2 protein causing Charcot-Marie-Tooth disease type 4C affectitslocalization in the plasma membrane and endocytic pathway.Hum MolGenet,2009,18(23): 4603–4614.
[27]Stendel C,Roos A,Kleine H,Arnaud E,Özçelik M, Sidiropoulos PNM,Zenker J,Schüpfer F,Lehmann U, Sobota RM,Litchfield DW,Lüscher B,Chrast R,Suter U, Senderek J.SH3TC2,a protein mutant in Charcot-Marie-Tooth neuropathy,links peripheral nerve myelination to endosomal recycling.Brain,2010,133(8):2462–2474.
[28]Kalaydjieva L,Nikolova A,Turnev I,Petrova J,Hristova A, IshpekovaB,PetkovaI,ShmarovA,Stancheva S, Middleton L,Merlini L,Trogu A,Muddle JR,King RH, Thomas PK.Hereditary motor and sensory neuropathy——Lom,a novel demyelinating neuropathy associated with deafness in gypsies.Clinical,electrophysiological and nerve biopsy findings.Brain,1998,121(3):399–408.
[29]Kalaydjieva L,HallmayerJ,ChandlerD,Savov A, Nikolova A,Angelicheva D,King RHH,Ishpekova B, Honeyman K,Calafell F,Shmarov A,Petrova J,Turnev I, Hristova A,Moskov M,Stancheva S,Petkova I,Bittles AH,Georgieva V,Middleton L,Thomas PK.Gene mapping in Gypsies identifies a novel demyelinating neuropathy on chromosome 8q24.Nat Genet,1996,14(2):214–217.
[30]Pietiäinen V,Vassilev B,Blom T,Wang W,Nelson J, Bittman R,Bäck1 N,Zelcer N,Ikonen E.NDRG1 functions in LDL receptortrafficking by regulating endosomal recycling and degradation.J Cell Sci,2013, 126(17):3961–3971.
[31]Warner LE,Mancias P,Butler IJ,McDonald CM,Keppen L,Koob KG,Lupski JR.Mutations in the early growth response 2(EGR2)gene are associated with hereditary myelinopathies.Nat Genet,1998,18(4):382–384.
[32]Warner LE, Svaren J, Milbrandt J, Lupski JR. Functional consequences of mutations in the early growth response 2 gene(EGR2)correlate with severity of human myelinopathies.Hum Mol Genet,1999,8(7):1245–1251.
[33]Topliko P,Schneider-Maunoury S,Levi G,Baron-Van Evercooren A,Chennoufi ABY,Seitanidou T,Babinet C, Charnay P.Krox-20 controls myelination in the peripheral nervous system.Nature,1994,371(6500):796–799.
[34]Decker L,Desmarquet-Trin-Dinh C,Taillebourg E, Ghislain J,Vallat JM,Charnay P.Peripheral myelin maintenance is a dynamic process requiring constant Krox20 expression.J Neurosci,2006,26(38):9771–9779.
[35]EspinósC,CalpenaE,Martínez-Rubio D,Lupo V. Autosomalrecessive Charcot-Marie-Tooth neuropathy. Adv Exp Med Biol,2012,724:61–75.
[36]Guilbot A,Williams A,Ravisé N,Verny C,Brice A, Sherman DL,Brophy PJ,LeGuern E,Delague V,Bareil C, Mégarbané A,Claustres M.A mutation in periaxin is responsible for CMT4F,an autosomal recessive form of Charcot-Marie-Tooth disease.Hum Mol Genet,2001, 10(4):415–421.
[37]Williams AC,Brophy PJ.The function of the Periaxin gene during nerve repair in a model of CMT4F.J Anat, 2002,200(4):323–330.
[38]Sevilla T,Martínez-Rubio D,Márquez C,Paradas C, Colomer J,Jaijo T,Millán JM,Palau F,Espinós C.Genetics of the Charcot-Marie-Tooth disease in the Spanish Gypsy population: the hereditary motor and sensory neuropathy-Russe in depth. Clin Genet, 2013, 83(6):565–570.
[39]Hantke J,Chandler D,King R,Wanders RJA,Angelicheva D,Tournev I,McNamara E,Kwa M,Guergueltcheva V, Kaneva R,Baas F,Kalaydjieva L.A mutation in an alternative untranslated exon of hexokinase 1 associated with hereditary motorand sensory neuropathy-Russe (HMSNR).Eur J Hum Genet,2009,17(12):1606–1614.
[40]Delague V,Jacquier A,Hamadouche T,Poitelon Y,Baudot C,Boccaccio I,Chouery E,Chaouch M,Kassouri N, Jabbour R,Grid D,Mégarbané A,Haase G,Lévy N. Mutations in FGD4 encoding the Rho GDP/GTP exchange factor frabin cause autosomal recessive Charcot-Marie-Tooth type 4H.Am J Hum Genet,2007,81(1):1–16.
[41]Horn M,Baumann R,Pereira JA,Sidiropoulos PNM, Somandin C,Welzl H,Stendel C,Lühmann T,Wessig C, Toyka KV,Relvas JB,Senderek J,Suter U.Myelin is dependent on the Charcot-Marie-Tooth Type 4H disease culprit protein FRABIN/FGD4 in Schwann cells. Brain,2012,135(12):3567–3583.
[42]Chow CY,Zhang YL,Dowling JJ,Jin N,Adamska M, Shiga K,Szigeti K,Shy ME,Li J,Zhang XB,Lupski JR, Weisman LS,Meisler MH.Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J.Nature,2007,448(7149):68–72.
[43]Nicholson G,Lenk GM,Reddel SW,Grant AE,Towne CF, Ferguson CJ,Simpson E,Scheuerle A,Yasick M,Hoffman S,Blouin R,Brandt C,Coppola G,Biesecker LG,Batish SD,Meisler MH.Distinctive genetic and clinical features of CMT4J:a severe neuropathy caused by mutations in the PI(3,5)P2phosphatase FIG4.Brain,2011,134(7):1959–1971.
[44]Chow CY,Landers JE,Bergren SK,Sapp PC,Grant AE, Jones JM,Everett L,Lenk GM,McKenna-Yasek DM, Weisman LS,Figlewicz D,Brown RH,Meisler MH. Deleterious variants of FIG4, a phosphoinositide phosphatase,in patients with ALS.Am J Hum Genet,2009, 84(1):85–88.
[45]Lenk GM,Ferguson CJ,Chow CY,Jin N,Jones JM,Grant AE,Zolov SN,Winters JJ,Giger RJ,Dowling JJ,Weisman LS,Meisler MH.Pathogenic mechanism of the FIG4 mutation responsibleforCharcot-Marie-Tooth disease CMT4J.PLoS Genet,2011,7(6):e1002104.
[46]Dubourg O,Azzedine H,Verny C,Durosier G,Birouk N, Gouider R,Salih M,Bouhouche A,Thiam A,Grid D, Mayer M,Ruberg M,Tazir M,Brice A,LeGuern E. Autosomal-recessive forms of demyelinating Charcot-Marie-Tooth disease.Neuromolecular Med,2006,8(1–2):75–86.
(责任编委:夏昆)
Advances in genetic studies of Charcot-Marie-Tooth disease type 4 (CMT4)
Ye Xu1,Jiaying Zhang1,BoyuYang1,Zhihong He1,Muchen Zhang1,ZhenYu2,Mingmin Gu2
1.School of Medicine,Shanghai Jiao Tong University Shanghai 200025,China;
2.Department of Medical Genetics,Shanghai Jiao Tong University School of Medicine,Shanghai 200025,China
The Charcot-Marie-Tooth disease(CMT)is one of the most common human inherited peripheral neuropathies.The most common pattern of inheritance is autosomal dominant,with less often occurrence autosomal recessive and X-linked dominant/recessive inheritance.CMT is generally divided into three forms:demyelinating forms(CMT1),axonal forms(CMT2)and intermediate forms(DI-CMT).The autosomal recessive form(AR-CMT1 or CMT4)is accompanied by progressive distal muscle weakness and atrophy of the limbs,pes cavus and claw-like hands.In addition,CMT4 is also characterized by early onset,rapid progression,and varying degrees of sensory loss and spinal deformities(e.g.scoliosis).Recently,11 subtypes of CMT4 have been identified.Some of these subtypes were clear in pathogenic mechanisms,some had founder mutation,but some still had limited clinical description and mutation analysis.In this review,we summarize the latest research progresses of CMT4,including genotypes and phenotypes,pathogenic mechanisms and mouse models.
Charcot-Marie-Tooth disease;CMT type 4;pathogenic mechanism;mouse model
URL:http://www.cnki.net/kcms/detail/11.1913.R.20150420.1357.001.html
2014-11-25;
2015-03-25
国家自然科学基金项目(编号:30470951,31071107)资助
许烨,临床医学专业八年制学生,专业方向:临床医学。E-mail:136965680@qq.com
顾鸣敏,教授,硕士生导师,研究方向:遗传病的基因定位和功能研究。E-mail:gumm@sjtu.edu.cn
10.16288/j.yczz.14-412
时间:2015-4-20 13:57:36
猜你喜欢
中华耳科学杂志(2022年1期)2022-11-24
磁共振成像(2022年8期)2022-10-08
中国生殖健康(2020年2期)2021-01-18
中成药(2019年12期)2020-01-04
党的生活(黑龙江)(2018年9期)2018-10-17
中成药(2018年7期)2018-08-04
小学生导刊(2018年13期)2018-06-29
益寿宝典(2018年1期)2018-01-27
中成药(2017年12期)2018-01-19
中国生殖健康(2018年2期)2018-01-12
