
线粒体脑肌病29例临床特点分析
2015-09-26 02:17:14邢海辉余年徐辉姚燕林兴建
神经损伤与功能重建 2015年4期
邢海辉 ,余年 ,徐辉 ,姚燕 ,林兴建
线粒体脑肌病29例临床特点分析
邢海辉1,余年2,徐辉2,姚燕2,林兴建2
目的:分析线粒体脑肌病(ME)患者各种非特异性临床表现和辅助检查结果,为该病早期诊断提供思路。方法:回顾性分析我院2008年05月至2013年12月收治的29例ME患者临床表现,寻找ME相对特异性的症状组合,并探索各项辅助检查结果(血和脑脊液的乳酸水平、脑电图、肌电图、神经影像、肌肉活检和基因检测)对各不同类型ME诊断的意义。结果:①本组患者最常见的临床表现依次为:癫痫(89.70%)、头痛(75.90%)、发作性无力(65.50%)等。②血空腹乳酸或乳酸运动试验阳性24例(82.8%);22例患者行脑脊液检查,其中蛋白升高12例(54.5%),乳酸升高8例(36.4%)。③29例患者的头颅MRI显示颞叶受累26例(89.7%),枕叶受累23例(79.3%),顶叶受累23例(79.3%),额叶受累6例(20.7%),小脑受累2例(6.9%)。头颅磁共振波谱成像检查16例,病灶区均见明显乳酸峰,NAA峰下降。④22例患者行肌肉活检,有阳性发现14例(63.6%)。9例患者行基因检查,5例(55.6%)发现线粒体DNA 3243A>G点突变,1例(11.1%)发现线粒体DNA8344A>G点突变。结论:ME为一种具有特殊影像学及病理表现的疾病,临床表现为多组非特异性症状的组合,易误诊,肌肉活检及基因检测可为最终确诊提供重要的依据。
线粒体脑肌病;磁共振成像;肌肉活检;基因检测
线粒体脑肌病(Mitochondrial encephalopathy,ME)是一组由线粒体DNA或核DNA缺陷导致线粒体结构和功能障碍、ATP合成不足所致的以脑和肌肉受累为主的多系统疾病,其共同特征为轻度活动后即感到极度疲乏无力,休息后好转,肌肉活检可见破碎红纤维。1984年Pavlakis等[1]首先报道6例ME伴高乳酸血症和卒中样发作(mitochondrialencephalomyopathy,lactic acidosis,and stroke-like episodes,MELAS)患者,主要表现为卒中样起病、癫痫发作、乳酸酸中毒、肌肉无力等。由于其临床表现的非特异性,大多数患者难以早期确诊,易误诊为其他神经系统疾病,如原发性癫痫、脑梗死、脑炎等,以至于延误诊断[2]。现回顾性分析我院收治的ME患者29例的临床资料,以提高对此病的认识,为该病诊断与鉴别诊断提供依据。
1 资料与方法
1.1 一般资料
收集2008年5月至2013年12月我院所有出院诊断为ME的患者29例,男17例,女12例;平均年龄28岁,其中10~20岁12例,21~30岁7例,>30岁10例。所有肌肉活检和取静脉血均获得患者或家属的知情同意。
1.2 方法
1.2.1 血乳酸测定 应用氧化酶法测定运动前和运动后10min乳酸量。
1.2.2 脑电图检查 根据国际10/20系统放置头皮电极,以双耳为参考电极。诊断标准分为正常、轻度异常、中度异常及重度异常。
1.2.3 肌电图检查 取股四头肌、腓肠肌、胫前肌、三角肌、肱二头肌、拇短展肌、小指展肌等处检测,观察安静状态下正相电位、纤颤电位、束颤电位及轻收缩和大力收缩时肌电图表现。
1.2.4 影像学检查 头颅MRI检查,使用GESigna3.0T超导型磁共振扫描仪。头颅MRI增强检查,采用0.1~0.2mmol/kg的钆喷替酸葡甲铵(Gd-DTPA)造影剂。
1.2.5 肌肉活检检查 取股四头肌组织,一部分经液氮-异戊烷速冻处理,冰冻切片,7~8μm薄切,做苏木素-伊红(HE)、改良Gomori三色、ATP酶、还原酶辅酶I四唑氮还原酶、糖原、油红O染色、琥珀酸脱氢酶、细胞色素氧化酶C染色,普通光镜观察;另一部分标本经4%戊二醛,锇酸固定,树脂包埋,半薄切片定位后行超薄切片,铅-铀双染,H-800透射电镜观察。
1.2.6 基因检测 部分患者及家属知情同意后留取静脉血3m L及晨尿400m L送到南京鼓楼医院神经分子诊断室采用基因测序法进行基因检测。
2 结果
2.1 临床表现
本组29例ME患者,主要临床表现为以癫痫发作(26/29)、头痛(22/29)、发作性无力(19/29)、智能障碍(18/29)、发育异常(13/29)为主,其中部分病例合并视觉症状、眼外肌麻痹、听力下降、共济失调及其他系统受累(除脑和肌肉外)。
2.2 辅助检查结果
2.2.1 血乳酸测定及脑脊液检查 血空腹乳酸或乳酸运动试验阳性24例(82.8%)。22例患者行脑脊液检查,其中蛋白升高12例(54.5%),乳酸升高8例(36.4%)。
2.2.2 脑电图 29例患者均行脑电图检查,其中中重度异常18例(62.1%),轻度异常8例(27.6%),正常3例(10.3%)。
2.2.3 肌电图 行肌电图检查26例,其中神经源性损害12例(46.2%),肌源性损害8例(30.8%),混合性损害2例(7.7%),正常4例(15.4%)。
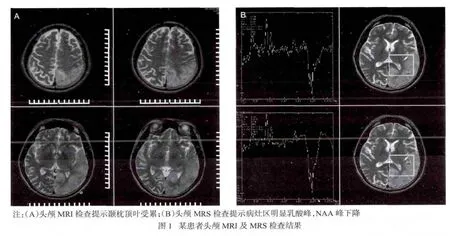
2.2.4 影像学检查 29例患者均行头颅MRI检查,颞叶受累26例(89.7%),枕叶受累 23例(79.3%),顶叶受累 23例(79.3%),额叶受累 6例(20.7%),小脑受累2例(6.9%)。前3者出现频率无明显差异,常以颞枕叶、顶枕叶或颞顶枕的组合方式同时受累,见图1A。头颅MRI增强检查22例,仅6例患者出现条索状强化。29例患者行头颅MRA检查,均未见明显异常。头颅磁共振波谱成像(magnetic resonance spectroscopy,MRS)检查 16 例,MRS病灶区均见明显乳酸峰,NAA峰下降,见图1B。
2.2.5 肌肉活检 22例患者行肌肉活检,发现破碎红纤维(RRF)和(或)琥珀酸脱氢酶深染肌纤维(RBF)和(或)电镜发现线粒体异常增多伴异常线粒体14例(63.6%),活检结果未见明显异常8例(36.4%)。
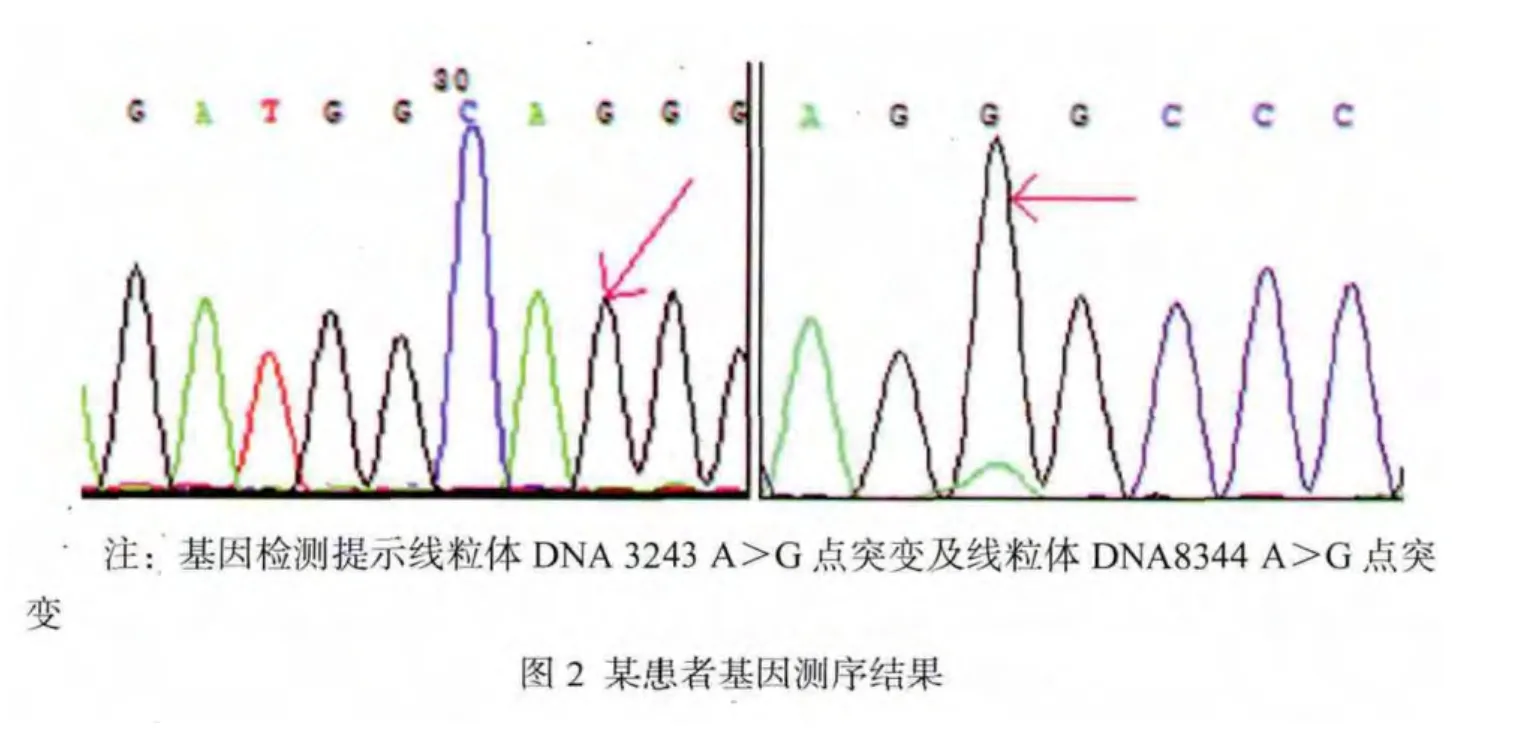
2.2.6 基因检测 9例患者行基因检测,5例(55.6%)发现线粒体DNA 3243 A>G点突变,1例(11.1%) 发现线粒体DNA8344 A>G点突变,见图2。3例(33.3%)基因检测未发现常见位点突变。
3 讨论
ME的临床表现复杂且缺乏特异性,常易误诊为癫痫、脑炎及脑梗死等其他脑病[3]。本组29例患者中,病初有12例误诊为病毒性脑炎,1例误诊为肝豆状核变性,10例拟诊为症状性癫痫,1例拟诊为脑占位性病变,仅5例在初诊时考虑ME。目前ME尚缺乏统一的诊断标准,确诊的金标准是在病理、基因和生化检查中发现线粒体有结构、分子和功能异常[4]。因为MELAS综合征相对常见,根据Yatsuga等[5]提出的MELAS诊断标准:①卒中样发作临床表现:头痛及呕吐;癫痫;偏瘫;皮质盲或偏盲;头颅CT或MRI显示相关的急性病灶。②线粒体功能障碍:血清乳酸升高或脑脊液乳酸升高;肌肉活检改良Gomori三色法染色可见破碎样红纤维(RRF)和(或)琥珀酸脱氢酶染色可见强血管反应,电镜示肌纤维细胞色素C氧化酶障碍或异常线粒体;MELAS相关基因突变。符合①项2条及②项2条者为确诊病例。在本组病例中,20例患者经肌肉活检病理证实线粒体结构异常和(或)基因检查发现m tDNA A3243G位点突变,证实线粒体分子水平异常,因此为确诊病例。其余9例因各种原因不能完善肌肉活检或基因检测,但是均出现血清乳酸升高或脑脊液乳酸升高,只能为临床诊断。ME主要包括4型,但临床上以MELAS型为主,因此笔者主要对于该型进行分析。
MELAS是以脑和骨骼肌症候为主的多系统受累疾病,其中多数患者以癫痫发作为其主要的起病形式,比例约为45%[6],开始多为单纯部分性发作,逐渐进展为全身性发作,以强直-阵挛发作最常见,常缺乏其他临床表现。患者一旦出现癫痫症状,往往会选择就医,故大部分患者以癫痫就诊。同时头痛作为一种潜在的神经功能障碍,也是MELAS的一种常见的临床特点,77%的MELAS患者可有偏头痛或其他形式的头痛,15%的患者头痛是其临床首发症状[7]。另外也常表现为认知障碍,在幼儿可表现为记忆力下降、学习能力差。肌病是MELAS患者典型的临床症状之一,而成人患者则多表现为发作性的肢体无力和运动不能耐受等[8]。根据对本组患者分析,MELAS患者多以青少年多见,首发症状主要为癫痫及头痛,也可表现为偏瘫、视野缺损、运动不耐受等,有时可伴有发育异常,如身材矮小、神经性耳聋等。上述临床症状反复发作,可逐渐出现智力下降、痴呆。癫痫发作包括部分感觉性发作、部分运动性发作、全身强直-阵挛发作,甚至出现癫痫持续状态。一般ME导致的癫痫应用丙戊酸钠后发作有加重趋势,临床选择药物应避免选用丙戊酸钠。针对患者表现为部分发作性的特点,可考虑选择卡马西平作为首选治疗药物,必要时加用新型抗癫痫药物联合治疗以控制临床癫痫发作。头痛系典型的偏头痛样发作,偏头痛伴有呕吐是MELAS典型的临床表现之一。本组患者中头痛常反复发作,以颞部搏动样疼痛最常见,常同时伴有发热,或癫痫发作、卒中样发作同时出现。卒中样发作是MELAS的重要临床表现之一,目前认为MELAS的卒中样发作并非血管源性,而是代谢引起的脑水肿,卒中样发作病灶分布范围常超过血管供血范围[9]。本组患者中出现卒中样发作最常见的是发作性偏瘫或单瘫,而偏盲相对少见,似乎与头颅MRI所见枕叶最常受累不相符,可能与偏盲容易被患者忽视有关。因此在发现患者出现上述症状时要考虑ME的可能。
MELAS的基本病理改变是脑组织海绵状变性、神经元变性或消失、星形胶质细胞增生及继发性脱髓鞘和铁沉积。这些改变并非特异,MELAS的病变主要累及各脑叶的脑回处,特别是多位于颞、顶和枕叶,但不按血管分布,以灶状坏死或软化为主,可见小血管异常增多,增生血管的管腔大小不等,管壁厚薄不均[10]。本组患者的头颅MRI主要特征为:①大脑、小脑半球皮质及皮质下白质病变,多个脑叶联合受累,主要以顶叶、颞叶及枕叶受累为主,少数累及小脑及额叶。病灶呈斑片状或楔形的脑梗死样病灶,边界欠清,并可同时累及大脑深部灰质和大脑皮质。②病变累及范围较大,不按大脑动脉供血区分布。③MRI信号欠均匀,T1WI像上多呈等、低信号,T2W I像为高信号。④急性期病变区域脑组织轻度肿胀,可有轻微或无占位效应。⑤头颅MRI增强无强化或轻度强化。⑥头颅MRA一般无异常。⑦MRS检查可在有信号异常的病变区及无信号异常的脑区(包括脑室系统)检测到具有一定特征的乳酸峰。MRS能早于形态学的改变提示诊断ME,目前的观点认为ME代谢变化早于形态学的变化。Abe等[11]报道在对MELAS患者的研究中发现,利用MRS检出乳酸双峰的时间比DWI异常高信号的时间要早2周左右。因此MRS可能有助于ME的早期诊断和对病理生理机制的理解,临床上如果高度怀疑ME,磁共振常规和DWI未见明显异常信号,MRS如果检测到异常的乳酸峰,则有助于ME的诊断。
对本病的诊断单纯依靠临床表现诊断是不可靠的。本组24例患者首次就诊时误诊,后经肌肉活检或基因检测后明确诊断。肌肉活检是诊断MELAS的重要方法,任何年龄段的肌肉活检发现多于2%的肌纤维出现RRF被认为是诊断MELAS的病理标准[12]。另外基因突变分析是确诊MELAS的重要手段。自1990年确立了MELAS的基因诊断以来,发现MELAS主要是m tDNA突变,其突变位点很多,可位于 tRNA、COXIII、NDl和ND5[13]。而发现最早、最常见的突变是编码tRNALeu的m tDNA A3243G突变,占80%左右[14]。其次为 tRNALeu3271T>C,约占7%~15%[15]。鉴于其基因突变异质性,对于A3243G突变检测阴性者,临床并不能排除MELAS,尚需增加基因检测位点以防漏诊,在本组病例中有3例患者行基因检测后未发现该位点突变。因此对于临床上高度怀疑ME的患者,应积极建议患者完善肌肉活检及基因检测。
对于ME的治疗,目前许多研究提示“鸡尾酒”疗法对改善患者症状和预后有一定疗效,其中包括维生素B1、维生素B2、维生素 B6、维生素 B12、维生素 C、维生素 E、辅酶 Q10、左卡尼汀、α-硫辛酸等,本文部分患者经“鸡尾酒”疗法对症治疗后症状得到明显的改善。然而,上述治疗方法均缺乏很好的临床对照试验,需要更为科学严谨的临床试验进一步证实各种治疗措施的确切效果[16]。目前为止仅有l项随机双盲临床试验证实肌酸、辅酶Q10、硫辛酸联合治疗可降低ME患者静息状态下的乳酸水平,减轻氧化应急反应[17]。最近有研究[18]报道改良“鸡尾酒”疗法,即在传统的“鸡尾酒”方案中加入丁苯酞,治疗8例ME患者,结果表明ME患者临床症状获不同程度的改善;但由于该研究样本量较少且部分患者依从性较差,其确切疗效需扩大样本进一步研究。另外积极控制癫痫发作,对症治疗头痛,控制和预防发热有助于降低脑组织的代谢水平,减少乳酸堆积,对于改善患者的预后至关重要。
ME作为一种具有特殊影像学及病理表现的疾病,临床表现多组非特异性症状的组合,易误诊,应引起临床医师的重视。临床上出现癫痫、头痛、发作性无力,不能耐受疲劳的患者,要警惕ME的可能,需尽早完善乳酸试验、磁共振检查,有条件的医院可行肌肉活检及基因检测进一步确诊。
[1]Pavlakis SG,Phillips PC,DiMauro S,et al.Mitochondrialmyopathy,encephalopathy,lactic acidosis and,stroke-like episodes:a distinctive clinical syndrome[J].Ann Neurol,1984,16:481-488.
[2]陈和成,杨金升,石向群,等.线粒体脑肌病的临床、病理与神经电生理[J].卒中与神经疾病,2007,14:28-31.
[3]Sheerin F,Pretorius PM,Briley D,et a1.Differential diagnosis of restricted diffusion confined to the cerebral cortex[J].Clin Radiol,2008,63:1245-1253.
[4]袁云.线粒体脑肌病伴高乳酸血症和卒中样发作的临床研究进展 [J].中华神经科杂志,2007,53:775-776.
[5]Yatsuga S,Povalko N,Nishioka J,et al.MELAS:A nationwide prospective cohort study of96 patientsin Japan[J].Biochim BiophysActa,2012,1820:619-624.
[6]Barshop BA,Naviaux RK,M cGowan KA,et al.Chronic treatment of Mitochondrial disease patients with dichloroacetate [J].Mol Genet Metab,2004,83:138-149.
[7]Sproule DM,Kaufmann P,Engecstad K,etal.Wolff-Parkinson-White syndrome in patients with MELAS [J].Arch Neurol,2007,64:1625-1627.
[8]Van Adel BA,Tamopolsky MA.Metabolic myopathies:update 2009[J].JClin Neuromuscul Dis,2009,10:97-121.
[9]熊葶,李尊波,罗国刚,等.线粒体脑肌病伴高乳酸血症和卒中样发作综合征一例报告[J].神经损伤与功能重建,2014,9:358-360.
[10]陈莉,卢德宏.线粒体脑肌病的神经病理学[J].卒中与神经疾病,1998,5:173-176.
[11]Abe K,Yoshimura H,Tanaka H,et al.Comparison of conventional and diffusion-weighted MRI and proton MR spectroscopy in patients with mitochondrial encephalomyopathy. Lactic aciosis, and stoke-like events[J].Neuroradiology,2004,46:113-117.
[12]姚生,戚晓昆.MELAS综合征 [J].中国神经免疫学和神经病学杂志,2010,17:228-231.
[13]Finsterer J. Central nervous system manifestations of Mitochondrial disorders[J].Acta NeurolScand,2006,114:217-238.
[14]Choi BO,Hwang JH,Cho EM,et al.MutationalanalysisofwholeMitochondrialDNA in patientswith MELASand MERRFdiseases[J].Exp MolMed,2010,42:446-455.
[15]Stenqvist L,Paetau A,Valanne L,et al.A juvenile case of MELAS with T3271C Mitochondrial DNA mutation[J].Pediatr Res,2005,58:258-262.
[16]Kerr DS.Treatment of mitochondrial electron transport chain disorders:a review of clinical trialsover the pastdecade[J].MolGenet Metab,2010,99:246-255.
[17]Rodriguez MC,MacDonald JR,Mahoney DJ,et al.Beneficial effects of creatine,CoQl0,and lipoic acid in Mitochondrial disorders[J].Muscle Nerve,2007,35:235-242.
[18]刘建国,姚生,李长青,等.改良"鸡尾酒"疗法对线粒体脑肌病的疗效观察[J].中国神经免疫学和神经病学杂志,2011,1:31-36.
Analysis on the Clinical Characteristics of Twenty-nine Patien ts with Mitochond rial Myopathy
XING Hai-hui,YU Nian,XU Hui,YAO Yan,LIN Xing-jian.Department ofNeurology,Nanjing Gaochun TraditionalChinese Hospital,Nanjing 211300,China
Objective:To analyze a variety of non-specific clinical features and laboratory exam ination results of mitochondrialmyopathy(ME)patients,and to provide information for the early diagnosisof this disease.Methods:A retrospective study was performed to analyze the clinicalmanifestations of 29 patientswith ME atour hospital from May 2008 to December 2013.A combination of relatively specific symptoms was explored and the significance of the results of the laboratory exam inations[lactate levels in plasma and cerebrospinal fluid(CSF),EEG,EMG,MRI/MRS,muscle biopsies and genetic testing]on the diagnosis of different types of ME was analyzed.Results:①The most common clinical manifestations in our patients included epilepsy (89.70%),headache(75.90%),weakness(65.50%)and the others.②Twenty-fourof 29 patients(82.8%)showed high fasting blood lactate levelor positive lactic exercise test.In the CSF exam ination,12 of 22 patients(54.5%)showed elevated CSF protein and 8 of the 22 cases(36.4%)had elevated CSF lactate.③All the patientswere given brain MRIscan and 26 of 29(89.7%)patientsshowed significant temporal lobe involvement.Additionally,therewere 23 cases(79.3%)with occipital lobe involvement,23 cases(79.3%)with parietal lobe involvement,6 cases(20.7%)with frontal lobe involvementand 2 cases(6.9%)with cerebellar involvement.The brainmagnetic resonance spectroscopy(MRS)exam inationwas completed in 16 cases,and allof their lesionswere apparent in lactate peak and NAA peak falling.④Twenty-two patients underwentmuscle biopsy.Therewere 14 cases(63.6%)with positive findings.Nine cases underwentgenetic testing,and five cases(55.6%)were foundMitochondrial DNA 3243 A>G pointmutation,and one cases(11.1%)was foundwithmitochondria DNA8344A>G pointmutation.Conclusion:ME had special radiologicaland pathologicalmanifestations,buthad themultiple combinations of clinical featureswithmany non-specific symptoms.ME is often easilym isdiagnosed.Muscle biopsy and genetic testing can provide an importantbasis for the definite diagnosis.
mitochondrialmyopathy;magnetic resonance imaging;muscle biopsy;genetic testing
R741;R741.04;R746
A
DOI10.3870/sjsscj.2015.04.007
1.南京市高淳中医院神经内科 南京211300
2.南京医科大学附属脑科医院神经内科 南京210029
南京市医学科技发展资金暨南京市卫生青年人才培养工程 (No.QRX11119)南京医科大学科技发展基金面上项目(No.2013NJMU09 0)
2015-03 -21
林兴建
linppmm@126.com
(本文编辑:唐颖馨)
猜你喜欢
海洋通报(2021年1期)2021-07-23 01:55:14
中国民间疗法(2021年5期)2021-06-09 09:21:04
生物学通报(2021年4期)2021-03-16 05:41:26
中华养生保健(2020年8期)2021-01-14 01:13:58
心肺血管病杂志(2020年3期)2021-01-14 00:42:38
饮食科学(2017年5期)2017-05-20 17:11:53
腹腔镜外科杂志(2016年12期)2016-06-01 12:10:09
河北科技大学学报(2015年6期)2015-03-11 16:16:51
西南军医(2015年4期)2015-01-23 01:19:30
癌变·畸变·突变(2014年1期)2014-03-01 04:39:36
