
两种功能化纳米金粒子固酶电极的催化性能
2015-09-15 01:40赵淑贤
无机化学学报 2015年12期
曾 涵 杨 阳 赵淑贤
(新疆师范大学化学化工学院,乌鲁木齐 830054)
酶燃料电池具有对环境友好和较高能量转化效率等优点,已成为当前的研究热点。研究者们普遍认为制约酶燃料电池性能的关键因素是阴极氧分子电催化还原过程[1]。漆酶(Laccase,缩写为Lac)因其活性中心具有较高的式电位,对氧分子具有较高的亲和力和高选择催化性能,被认为是酶燃料电池阴极的首选电化学催化剂,但Lac结构复杂,其活性中心为多肽链包覆,过去认为很难实现电子在酶-电极之间的直接迁移[2-3]。迄今为止已有多种方法实现Lac分子和电极之间的直接电子迁移[4-7],这些方法中最引入注目的就是利用纳米器件表面功能化特定结构官能团的方法,使氧化还原基团或与酶活性中心发生特殊相互作用的基团靠近酶活性中心,以利于酶活性中心-电极间的直接电子迁移,考虑到Lac活性中心结构的复杂性,通常使用的将葡萄糖氧化酶活性中心在电极表面重构的方法[8]对于Lac难以实现。近期许多文献[9-14]都指出,以具有大π共轭体系的芳香化合物修饰纳米器件可以利用π-π堆积作用使酶活性中心定向排列在纳米器件表面并与表面修饰的特定官能团发生特定相互作用,利于酶-导电基体间的直接电子迁移。纳米金粒子作为常用的固酶载体和构筑酶基电极的导电基体具有很多优点:导电性能优异、比表面积高、粒径可控以及表面修饰功能化基团简便等[15-16],但是纳米金粒子与酶分子直接接触时,可能因为纳米金粒子与酶活性中心附近多肽配体间的相互作用,造成酶活性中心构型扭曲,导致催化活性下降甚至失活[17]。目前已有一些文献报道采用芳香基团包覆或者修饰纳米金粒子表面的方法,构筑实现酶-电极间直接电子迁移[18-20],但仍有改进的空间:制备繁琐,成本高,时间长;由于只能固定单层或几层实现导电的酶分子,导致催化性能不高(有的固定Lac电极虽可实现酶-电极间直接电子迁移但对底物无催化性能[19]),以及使用致癌的稠环芳烃,难于在生物体内使用等。文献报道过2种不同官能团修饰纳米金粒子的制备方法[21],迄今为止尚未见其作为固酶载体并以之为基础,制备酶基电极以及测试其性能的研究报道,只有使用氧化还原电子中介体-酶自组装层叠层体系得到固酶电极并讨论其催化氧还原性能的报道[22]。基于上述思路,本文分别以这两种纳米金粒子作为固酶载体和导电基体,制备了4种纳米金粒子固酶修饰电极 (每一种纳米金粒子分别固定Lac和葡萄糖氧化酶GOx),本文提出的这类固酶载体,凭借纳米金粒子表面芳香环-酶分子活性中心周围疏水结合位间的疏水-疏水作用,使酶活性中心定向排列在纳米金粒子表面;同时依靠纳米金粒子表面官能团与酶分子表面氨基酸残基之间的相互作用,物理吸附或化学偶联酶分子,这样不仅将酶分子牢固地固定于电极表面,也利于实现酶-电极间的直接电子迁移。通过比较这两种不同结构纳米金粒子固酶电极的直接电子迁移和催化底物反应的性能,再结合前文和文献给出的类似固酶电极的测定结果,为定性和定量分析固酶方式、电极表面固酶载体结构和形貌等电极结构参数对固酶电极催化性能的影响提供了实验基础,结合相关理论,有可能深入分析和阐释生物体内神经信号,新陈代谢等生理活动的机制。本文还以这两种纳米金粒子构筑的固酶电极组装了2种酶燃料电池,测试了其能量输出性能,进一步评估了其长期使用性,确定了影响电池性能的主要因素。
1 实验部分
1.1 仪器和试剂
聚聚乙烯亚胺 (PEI,Mr:7.5×105), 聚乙烯基吡啶(PVP,Mr:6.0×104),聚丙烯酸(PAA,Mr:2.4×105),4-巯基苯甲酸(MBA,分析纯),巯基乙胺(分析纯),漆酶(Lac,来源于 Trametes Versicolor,Mr:6.8×104), 葡萄糖氧化酶(GOx,Mr:~5.0×104),2,2′-连氮-双-(-3-乙基苯并噻唑啉-6-磺 酸)-二 胺 盐 (ABTS,98.5%), 牛 血 清 白 蛋 白(BSA), 四水合四氯金酸,NaBH4,N-羟基琥珀酰亚胺(NHS,AR),N-乙基-N′-(3-二甲基氨丙基) 碳二亚胺(EDC,AR)均购自美国Sigma-Aldrich化学试剂有限公司,冰乙酸,甲醇,乙醚以及配制磷酸盐缓冲液(PBS)所需的试剂均为分析纯。
Bruker Equindx-55型红外光谱仪 (德国Bruker公司),KBr压片;透射电镜照片以H-800型透射电子显微镜(日本日立公司,加速电压:200 kV)拍摄;Analyst 800型原子吸收光谱仪(美国Perkin-Elmer公司,主机:双光束火焰/石墨炉原子吸收分光光度计,光谱范围:190~870 nm);U-2810型紫外可见分光光度计 (日本岛津公司,比色皿厚度1 cm);CHI-1140A型电化学分析仪(上海辰华仪器有限公司),作为参比电极的Ag/AgCl(饱和KCl)电极和作为基底电极的金盘电极(GD,直径1 mm)均购自天津艾达恒晟工贸有限公司,自制铂丝电极作为对电极,高纯氮气和高纯氧气购自南京特气,Clark型氧电极(英国Hansatech公司)。基底金盘电极使用前先以3500#砂纸,1.0和0.5μm氧化铝粉浆抛光,再用丙酮和三次重蒸水超声清洗各2次,每次2 min。
1.2 功能化纳米金粒子的合成
两种不同结构的功能化纳米金粒子的制备方法如文献[21]所述,简述如下:(1)将四氯金酸和4-MBA(物质的量之比为1∶3)共同溶解于35 mL甲醇/乙酸混合液(6∶1,V/V)中,快速搅拌条件下加入含 0.3 g NaBH4的15 mL甲醇溶液,加热回流30 min,产生的黑色分散相在冷却到室温后继续搅拌30 min。随后将分散相转移到离心管中,于8 000 r·min-1下离心沉降10 min,得到的黑色固体以乙醚冲洗3次,随后用干燥N2气流吹干待用。此黑色固体即为4-巯基苯甲酸功能化功能化纳米金粒子 (4-MBA@GNP);(2)分别制备PVP的甲醇溶液和四氯金酸的甲醇溶液,按照金属盐-吡啶单元的物质的量之比为1∶2的比例,快速搅拌条件下将两者共混10 min后,快速向溶液中加入NaBH4浓度为3.0 mg·mL-1的甲醇溶液,此时可以观察到溶液颜色由棕黄色迅速变为微粉红色,表明金纳米粒子已经产生,此外由于溶液中发生的均相还原反应会导致溶液pH值升高。在进一步搅拌30 min后,将得到金纳米溶胶液冷却到室温。其中获得的纳米金溶胶即为聚乙烯基吡啶包覆纳米金粒子(Py-GNP)。
1.3 固酶电极的制备
使用2种纳米金粒子作为固酶载体分别构筑力学性能稳定的固酶阴极和固酶阳极 (即较大剪切力作用于电极表面时,两种电极表面修饰的纳米金粒子都不会脱落,与电极接触的溶液的UV-Vis谱图中没有观察到文献[21]中出现的功能化纳米金粒子的特征吸收峰),现分别简述如下:
1.3.1 对巯基苯甲酸功能化纳米金粒子偶联酶基电极的制备
首先将事先预处理过的金盘电极浸泡于质量分数为0.5%的PEI水溶液中6 h便可得到-NH2功能化的金电极,随后将此电极转移到含PAA浓度为0.3 g·L-1的甲醇溶液中继续浸泡30 min,便可得到-COOH修饰的电极表面,取出电极转移至PVP的甲醇溶液中(浓度为 0.3 g·L-1)浸泡 30 min以饱和吸附一层 PVP。随后将吸附PVP的金电极浸泡在含4-MBA@GNP浓度为 0.5 g·L-1的甲醇-乙酸(10∶1,V/V)混合溶液中 1 h,获得对巯基苯甲酸功能化纳米金粒子修饰电极 (记为4-MBA@GNP/Au),再以EDC和NHS(物质的量之比为5∶1)的混合液活化此电极表面的羧基。最后将活化后的4-MBA@GNP/Au浸泡在含有GOx浓度为3.0 g·L-1或含有 Lac 浓度为 6.0 g·L-1的 0.2 mol·L-1PBS 缓冲溶液中 (pH=6.0)8 h,即可得到固酶阳极 GOx/4-MBA@GNP/Au和固酶阴极Lac/4-MBA@GNP/Au。类似地,在修饰一层酶分子后,按照之前相同的方法,此电极先后浸入PVP的甲醇溶液和4-MBA@GNP的甲醇-乙酸混合液后就可以获得未偶联酶的新4-MBA@GNP修饰电极表面,可以继续固定酶分子,重复这一过程,就可以得到自组装的层叠层纳米金粒子固酶电极。这种电极的构筑过程参见图1。
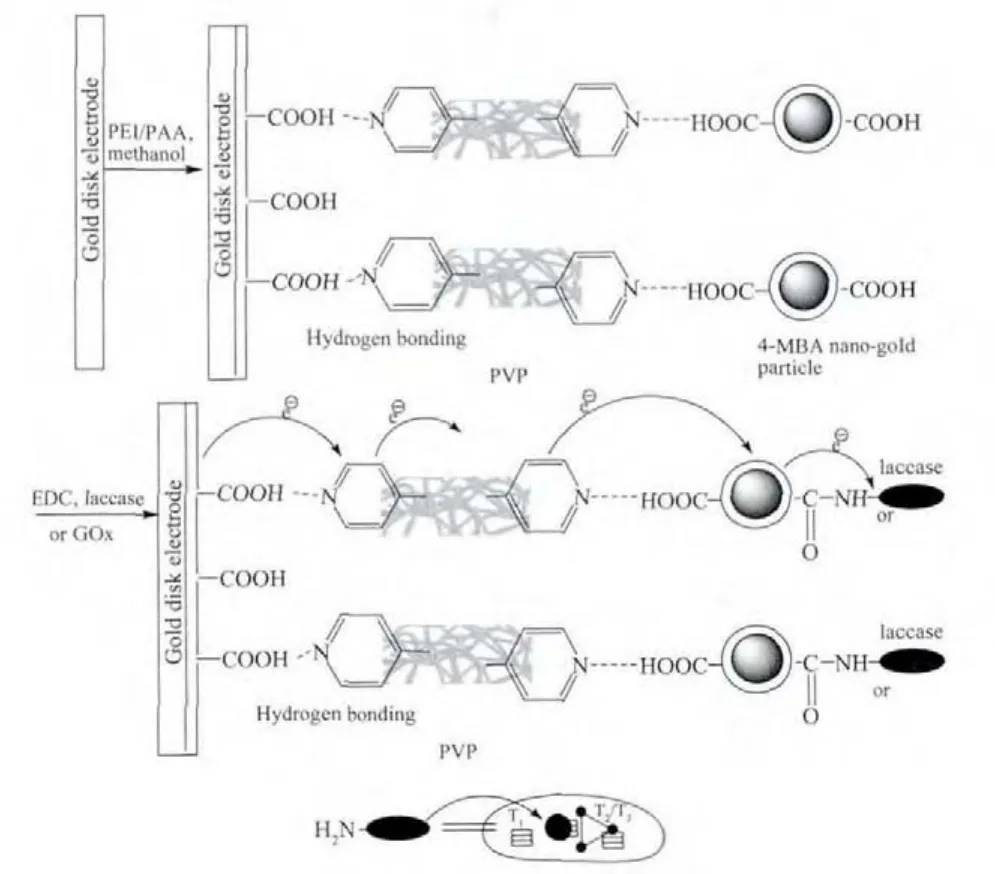
图1 对巯基苯甲酸功能化纳米金粒子固酶自组装电极制备示意图Fig.1 Schematic illustration for the preparation of 4-mercaptobenzoic acid functionalized nano-gold particle with immobilized enzymes self-assembly multi-layered electrode
1.3.2 吡啶官能团功能化纳米金粒子吸附酶基电极的制备
首先将事先预处理过的金盘电极浸泡于质量分数为1.0%的巯基乙胺溶液中8 h便可得到-NH2功能化的金电极表面,随后将此电极转移到含PAA浓度为0.3 g·L-1的甲醇溶液中继续浸泡45 min,便可得到-COOH修饰的电极表面,取出电极转移至含Py-GNP浓度为0.5 g·L-1的甲醇溶液中6 h,凭借Py-GNP纳米粒子表面的吡啶基团中的N原子和-COOH之间的氢键作用,获得吡啶官能团功能化纳米金粒子修饰电极(记为Py-GNP/Au),最后将此电极于4℃条件下浸泡在含有GOx浓度为3.0 g·L-1或含有Lac浓度为6.0 g·L-1的0.2 mol·L-1PBS缓冲溶液中(pH=6.0)12 h,凭借 Py-GNP纳米粒子表面的吡啶基团中的N原子与酶分子表面氨基酸残基中的-COOH之间的氢键作用吸附酶分子,即可得到固酶阳极GOx/Py-GNP/Au和固酶阴极Lac/Py-GNP/Au。这种电极的构筑过程参见图2。

图2 聚乙烯基吡啶包覆纳米金粒子固酶电极制备示意图Fig.2 Schematic illustration for the preparation of poly(vinylpyridine)overlappd nano-gold particle with entrapped enzymes modified electrode
1.4 固酶纳米金粒子的表征
纳米金粒子固酶前后的形貌以透射电镜(TEM)进行表征。采用紫外-可见分光光度法(UV-Vis)测定固定Lac纳米复合物吸收光谱的具体方法如下:将Lac溶解于 PBS 中得到 5.0×10-2g·cm-3的溶液(游离 Lac),倒入比色皿中进行紫外可见吸收光谱(UV-Vis)测定。将2种纳米金粒子溶液与酶充分接触(参看1.3节),分别制备好固定Lac的2种纳米金粒子复合物溶液 (浓度均为0.5 g·L-1),移取此液200μL均匀地涂覆在ITO玻璃片上,室温下干燥,最后插入比色槽中进行UV-Vis测定。
1.5 固酶电极的直接电化学及电催化性能
采用循环伏安法(CV)研究固酶电极的直接电化学行为及催化底物氧化/还原性能。将以上各种固酶阳极和固酶阴极分别作为工作电极,与参比电极(Ag/AgCl电极)以及自制对电极(铂丝)连接成一个三电极系统,置于一个容积为25 mL的玻璃电解池中。测试电极的直接电化学行为时,加入的电解质为不含任何底物且事先通入N2除氧至少30 min的PBS;测试固酶阳极的电催化氧化性能前,电解池内注入不同葡萄糖浓度的PBS缓冲液并以高纯N2鼓泡除氧至少30 min,测试时电解液上方继续通入N2以维持无氧气氛;而测试固酶阴极的电催化还原性能前,需向PBS缓冲液鼓泡通入高纯O2至少15 min以使溶液为O2饱和,测试时电解液上方继续通入O2以维持氧气气氛。文中给出的电流-电压曲线(i-E曲线)均为电极达到稳定状态时扫描所得 (即第五圈扫描时所得i-E曲线)。固酶电极担载酶的质量按照文献[23]给出的方法(用石墨炉原子吸收分光光度法测定电极固酶前后溶液中含铜量的变化,以计算电极表面固定Lac的质量)进行测定。
1.6 固酶燃料电池的组装和性能评估
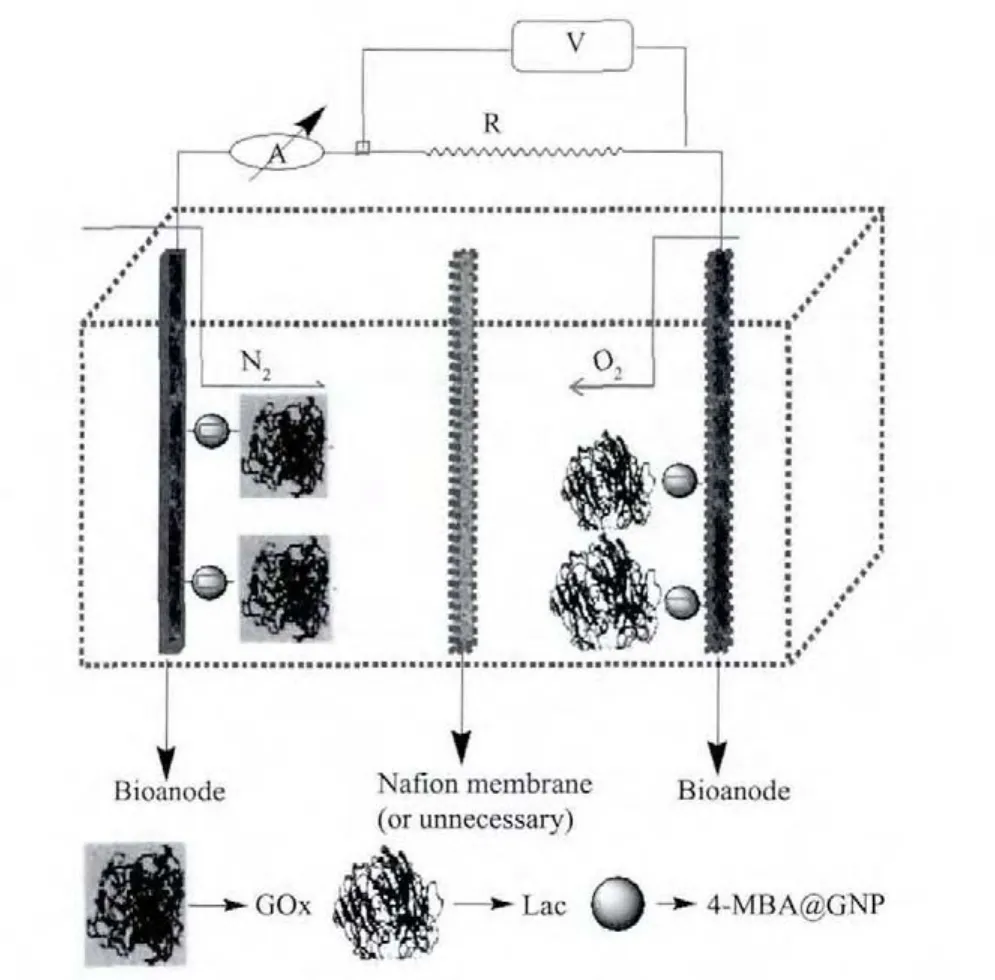
图3 组装的纳米功能金粒子固酶基燃料电池的示意图Fig.3 Schematic illustration for the fabrication of functionalized nano-gold particle with immobilized enzymes-based fuel cell
将构筑的固定GOx的阳极和固定Lac的阴极,放入一个玻璃制两极室电池中。2个极室上方各有一个进气口,以导气管与气源连接,阳极电解液含有不同浓度的除氧葡萄糖PBS缓冲溶液(pH=6.0);而阴极室则充入为氧气饱和的PBS缓冲溶液(pH=6.0),两者均无需加入任何电子中介体。电阻值范围在0~100 kΩ的外加负载与组装的两个电极相连接,籍调节负载的电阻值调控电池的输出电压和输出电流密度,电池的结构及组装示意图参见图3。
本文中如无特殊说明,所涉及的电位均为相对于标准氢参比电极 (NHE)的值。酶基阳极GOx/4-MBA@GNP/Au,GOx/Py-GNP/Au以及固酶阴极Lac/4-MBA@GNP/Au,Lac/Py-GNP/Au的活性表面积按文献[24]给出的方法:以铁氰化钾为探针,测定值分别为0.009、0.007 cm2和 0.011、0.010 cm2。测定的电流密度系输出电流对活性面积归一化所得。所有实验操作皆在室温(25.0±0.40)℃,常压下进行。
2 结果与讨论
2.1 固定Lac纳米金粒子的表征
将4-MBA@GNP与4-MBA的红外光谱FTIR进行比较后发现:前者的光谱图中除了2 556 cm-1附近对应于巯基SH伸缩振动吸收峰(参见支持信息图S1)消失之外,其他吸收峰位置与后者相似,这表明4-MBA成功地覆盖在纳米金粒子表面。图4为4-MBA@GNP固酶前(a)和后(b)的TEM照片(限于篇幅,仅以4-MBA@GNP固定Lac为例,4-MBA@GNP固定GOx,Py-GNP固定 GOx和 Lac的情况与此类似,其TEM不再给出)。从图4可以看出,制备的4-MBA@GNP纳米金粒子基本保持球形,其平均粒径大约15 nm;而4-MBA@GNP固定Lac后由于纳米金粒子表面固定酶分子官能团(-NH2和-COOH)之间相互作用-化学偶联以及氢键,导致纳米金粒子易于形成较大的无规则团簇,不再保持单个纳米金粒子的球形粒子形状,而且由于酶分子包覆于纳米金粒子的表面,使得固酶纳米金粒子团簇 (图4b)较单个纳米金粒子(图4a)的亮度更灰暗一些,这与文献[15]报道的类似体系结果相近。
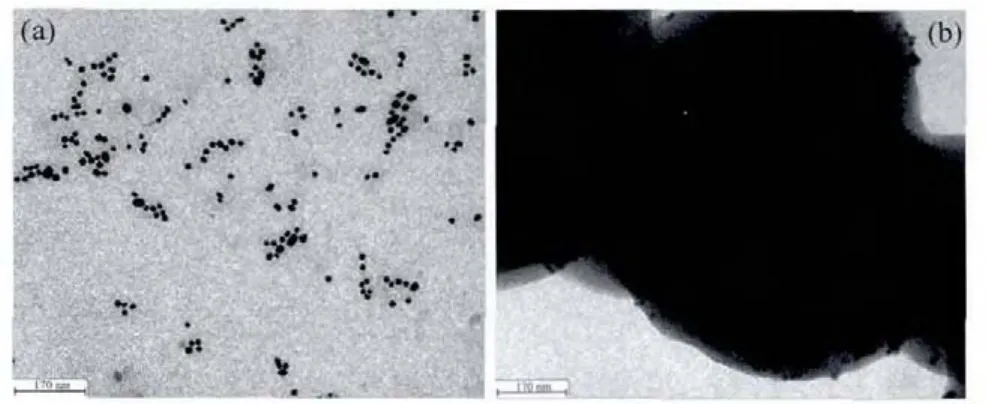
图4 4-MBA@GNP固定Lac前(a)和后(b)的TEM照片Fig.4 TEM images of 4-MBA@GNPbefore(a)and after(b)laccase immobilization
图5 为游离Lac溶液,Lac/4-MBA@GNP及 Lac/Py-GNP薄膜的UV-Vis光谱图,从图5可以看出:三者在~605 nm附近均出现一个强吸收峰,其对应于Lac活性中心T1氧化态铜离子在周围配体作用下的d-d配位跃迁[25]。这表明,2种功能化纳米金粒子固定的Lac均较好地保留游离Lac活性中心固有的配位构型和中心离子价态,但两者的吸收峰强度存在显著差异,由于4-MBA@GNP可通过化学偶联方式固定酶分子,不但可稳定地固定酶分子,同时也增加了纳米金粒子对Lac的担载量 (吸收峰强度明显高于Lac/Py-GNP);与此相对,Py-GNP仅是通过氢键作用固定Lac分子,不但稳定性较差,而且由于吡啶官能团在纳米金粒子表面分布的随机性,使其固定Lac量不高。石墨炉原子吸收法测定结果与上述结论相一致,Lac/4-MBA@GNP/Au和Lac/Py-GNP/Au对Lac的担载量分别为 240.0 和 56.0 mg·g-1。 同时测试结果还表明:石英片上固酶多层薄膜在400、500 nm(4-MBA@GNP固酶电极)以及530 nm(Lac/Py-GNP/Au固酶电极)处吸光度随固酶纳米金粒子修饰层数的增加线性升高,这一结果与文献[21]类似;此外605 nm处吸收峰强度会随着固酶层数的增加线性升高,这表明可以通过层叠层自组装的方式成功构筑固酶多层电极。

图5 游离Lac和2种功能化纳米金粒子薄膜的UV-Vis光谱Fig.5 UV-Vis spectra of free Lac solution,thin films of two kinds of functionalized nano-gold particles
2.2 固酶电极的直接电化学及催化氧还原性能
2.2.1 固酶电极的直接电化学行为

图6 固酶阳极GOx/4-MBA@GNP/Au(a)和固酶阴极Lac/4-MBA@GNP/Au(b)在无底物的除氧0.2 mol·L-1 PBS(pH=6.0)中扫描所得的CV曲线Fig.6 CV curves of bioanode GOx/4-MBA@GNP/Au(a)and biocathode Lac/4-MBA@GNP/Au(b)recorded in deaerated 0.2 mol·L-1 PBS(pH=6.0)in the absence of any substrate
图6 为固酶阳极GOx/4-MBA@GNP/Au以及自组装多层(5层Lac)固酶阴极Lac/4-MBA@GNP/Au分别在 0.2 mol·L-1无氧 PBS(pH=6.0)中扫描所得 CV 曲线(为了保证氧化还原峰电流之比数值接近且都接近于电子迁移可逆的状态,测试固酶阳极和固酶阴极在无底物和中介体的PBS溶液中CV曲线时采用不同电位扫描速率)。从图6a可以看出:,固酶前的纳米金粒子修饰金盘电极在扫描电位区间范围内没有任何与背景电流区分的氧化还原信号,而当该电极固酶之后,在同一电位区间范围内出现了一对明显的氧化还原峰,对应于GOx活性中心核黄素腺嘌呤二核苷酸(FAD)以准可逆方式得失电子的氧化还原峰(阴阳极峰电流之比 ip,a/ip,c=0.8,中值电位-244 mV,接近文献[26]报道的FAD式电位-220 mV vs NHE),按文献[15]介绍的方法可以估算出电极表面固定的、实现直接电子迁移的 GOx 分子浓度为 1.3×10-8mol·cm-2;而对于图 6b 所示的Lac/4-MBA@GNP/Au来说,则能观察到一对峰形更对称的氧化还原峰(ip,a/ip,c=0.86,峰电位差为67 mV,中值电位552 mV),根据文献[27]报道的Lac各活性中心的式电位数据,这对准可逆的氧化还原峰可归因于固定于电极表面的Lac活性中心T1与纳米金粒子间发生单电子氧化还原反应 (中值电位E1/2=780 mV vs NHE),电位负移230 mV应与纳米金粒子与Lac分子之间的相互作用(Au与酶分子表面氨基酸残基的相互作用)相关。根据前述估算方法也可以类似地估算出固酶阴极表面实现直接电子迁移的Lac分子浓度为1.24×10-8mol·cm-2, 大致与阳极表面实现直接电子迁移的GOx分子浓度相同,但仅有文献[28]报道的NAFION薄膜吸附Lac修饰热解石墨电极上导电酶分子浓度 6.6×10-8mol·cm-2的五分之一左右(需要说明的是,随着实现直接电子迁移的自组装固酶层数的增加,相同扫速下氧化还原峰面积及由此计算所得的导电酶分子浓度也随之线性升高,与2.1节结论一致,即表明电极表面实现直接电子迁移的酶分子以层叠层方式,成功构筑固酶多层电极)。
与 4-MBA@GNP/Au固酶电极不同,Py-GNP/Au固酶电极在不引入外加电子中介体时无法实现酶活性中心与导电基体-纳米金粒子间的直接电子迁移(限于篇幅,本文仅以Lac/Py-GNP/Au为例,参见图7和支持信息图S2,GOx/Py-GNP/Au与此类似,图不再给出),与文献报道的固酶电极类似,当加入电子中介体ABTS时才可有效实现酶-电极间电子迁移 (图7),出现一对ABTS氧化还原态相互转化的准可逆氧化还原峰,中值电位~720 mV,此反应受扩散控制[29]。分析Lac/4-MBA@GNP/Au在不含任何中介体的无氧PBS中以不同扫速获得的CV曲线(参见支持信息图S3)可知,无论是阳极峰电流还是阴极峰电流,在较低电位扫速范围(2~200 mV·S-1)内,峰电流与扫速保持良好的线性关系,而且峰电位基本保持稳定,ip,a/ip,c一直接近于1,这表明发生在此电极上的电化学反应是一个表面控制型的准可逆单电子氧化还原反应[15]。

图 7 固酶电极 Lac/Py-GNP/Au在含 ABTS1.0 mmol·L-1的无氧PBS(pH=4.4)中扫描所得的CV曲线Fig.7 CV curve of electrode with entrapped enzyme:Lac/Py-GNP/Au recorded in deaerated PBS(pH=4.4)in the presence of mediator at concentration level:1.0 mmol·L-1
2.2.2 固酶电极的催化氧还原性能
图8为2种不同结构的固酶阴极Lac/Py-GNP/Au(a)和Lac/4-MBA@GNP/Au(b)分别在含中介体ABTS浓度为1.0 mmol·L-1的PBS和不含任何中介体的 PBS中扫描获得的CV曲线。从图8a可以看出,Lac/Py-GNP/Au分别在无氧,空气饱和以及氧气饱和的PBS(pH=4.4)中扫描获得的CV曲线有明显区别:当溶液中含有氧气时,还原峰电流有明显的增加而氧化峰电流则明显的降低。此现象表明Lac在中介体存在时对氧还原有明显的电催化作用(参见支持信息图S2,当固酶电极在无中介体存在时对氧还原仅有极微弱的催化性能),可归因于氧气存在促进了酶催化氧化产生的ABTS阴离子自由基的氧化,从图8a还可以看出,这一电极对底物氧的浓度较为敏感,随着PBS中氧分压的增加,产生的催化氧还原电流也明显上升,但电极在氧气饱和及空气饱和的PBS中产生的催化电流之比(7.1)远高于 2 种溶液中氧气浓度之比(~4.6)。这表明催化循环的决速步并非是氧气扩散过程,而是ABTS氧化态在溶液中的扩散,这表明此催化反应主要受制于中介体的扩散过程。从图8b则可以看出,电极在氧气饱和溶液中所得CV中阴极还原电流在0.96 V开始急剧增加,并在0.8 V附近达到极限,同时阳极氧化峰消失,而且随着电极表面固定酶量的增加(表现为固酶层数的增多),催化还原电流随之上升,这些结果表明通过这种方式固定的Lac不但实现了酶活性中心T1与导电基体间的直接电子迁移,而且随着电极表面固酶量的增加,催化性能也得以进一步提升直至达到导电酶分子的饱和浓度,此时催化电流达到极限值(固酶自组装层数为5),此后继续增加电极表面固酶层数则催化电流将缓慢下降(限于篇幅,图不再给出)。这可以归因于过多的酶增大了电极表面的电阻,而且排列紧密的酶分子很可能由于分子间的相互作用聚集成簇(参见图4b),降低了酶分子与底物接触面积,导致酶催化活性下降。根据0.6 V时所得的极限催化电流ilim,cat=23.8μA,前面计算所得的导电Lac分子表面浓度Γ以及电极活性表面积A等数据,由文献[30]给出的公式可估算出实现酶-电极间直接电子迁移的Lac基电极上单位时间内氧分子转化平均速率为0.5 s-1,这一数值较文献[31,13,15]报道的类似电极的催化氧还原速率(0.14、0.09 和 0.06 s-1)有较大的提升(不同电位扫描速率下获得的还原电流密度虽然不一样,但是按照本文实验部分给出定义求算的极限催化电流密度以及不同扫速下求算的导电酶分子浓度却是相同的),表明以具有大π共轭体系修饰的纳米器件作为固酶载体的电极的确具有相对高的催化性能,但载体结构与催化效能的关系还需要进一步定量研究。
2.2.3 固酶阴极催化氧还原性能的重现性和长期稳定性
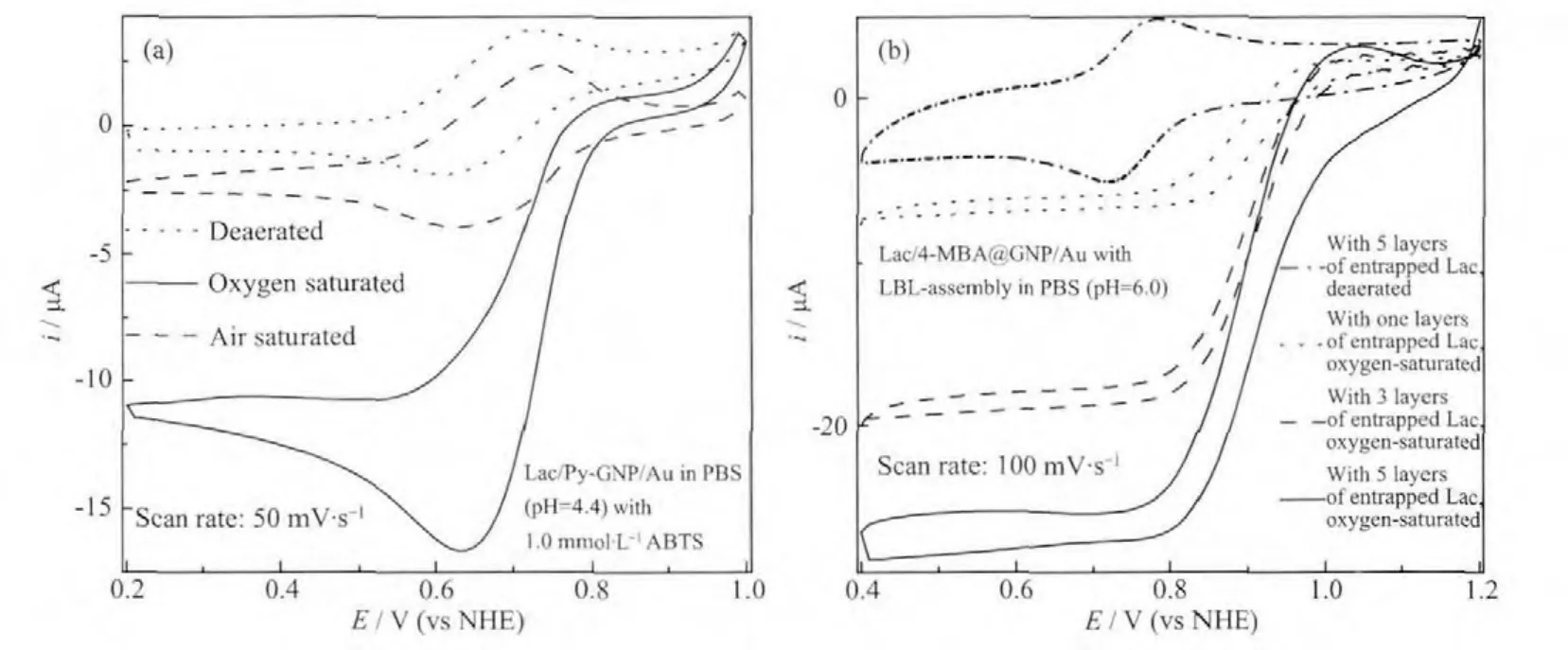
图8 CV法表征Lac/Py-GNP/Au(a)和Lac/4-MBA@GNP/Au(b)的催化氧还原性能Fig.8 Catalytic effect on oxygen reduction reaction(ORR)of Lac/Py-GNP/Au(a)and Lac/4-MBA@GNP/Au(b)characterized by CV
对于燃料电池而言,固酶电极的催化性能的重现性和长期稳定性是影响电池性能的重要指标和未来实现实用化的重要前提。支持信息图S4分别给出了5个不同时间按照同样方法制备的2种不同结构的固酶阴极(限于篇幅,固酶阳极测试结果与此类似,不再给出)在氧气饱和的PBS溶液中测得极限催化电流密度的对比图以及在同样溶液中所测得的极限催化电流密度与电极低温储存时间的依赖关系曲线,对于Lac/Py-GNP/Au而言,测试条件同图8a,而对于Lac/4-MBA@GNP/Au,测试条件参见图8b(自组装固酶层数为5)。从图S4可以看出,5个不同的Lac/4-MBA@GNP/Au所测得的极限催化电流密度没有明显差异,而相同数量的Lac/Py-GNP/Au之间的差异较前者更为显著,这表明Lac/4-MBA@GNP/Au催化性能重现性较Lac/Py-GNP/Au更佳。此外还可看出Lac/4-MBA@GNP/Au催化性能的稳定性也远优于Lac/Py-GNP/Au:前者在储存21 d后的极限催化电流密度仍然可以保持最初值的80%以上,而后者仅使用3 d后催化性能就降低到最初值的2/3左右,使用10 d后基本失去催化活性,以上结果均表明Lac/4-MBA@GNP/Au催化氧还原的重现性和长期使用性均远优于Lac/Py-GNP/Au,这除了后者固定Lac的稳定性不如前者之外,与中介体ABTS的长期使用稳定性能欠佳也有很大关联。
2.2.4 固酶阴极催化氧还原性能的热稳定性和酸碱耐受性
支持信息图S5给出了固酶阴极Lac/4-MBA@GNP/Au在氧气饱和的PBS溶液中 (测试条件参见图8b,自组装固酶层数为5),测得极限催化电流密度与温度的依赖关系曲线和极限催化电流密度-溶液pH值关系曲线,从图S5可看出:电极Lac/4-MBA@GNP/Au催化氧还原性能与游离Lac类似,存在一个最佳pH值(6.0)(游离Lac的最佳pH值为3.0附近,这种差异与载体-酶分子间的相互作用有关联[32-33]);还可观察到在测试温度范围内,随着温度升高,在328 K之前极限催化电流密度随温度升高而单调递增,而当温度超过328 K之后催化性能急剧下降,这个结果与文献[34]报道固酶电极的催化电流密度-温度关系非常相似。根据图中数据可以估算出电池催化反应的表观活化能大约是38.2 kJ·mol-1,而酶变性的活化能则大约是58.8 kJ·mol-1。以上结果表明此电极很可能以化学吸附的形式固定Lac,而当温度升高后酶变性从而造成电池输出性能下降,但较大的变性活化能也表明此电极对温度具有较强的耐受力。需要指出的是,另一种固酶阴极Lac/Py-GNP/Au催化氧还原的酸碱耐受性类似于文献[35]报道的固酶电极,均受限于固定酶固有的活性-pH值依赖关系和固酶载体对酸碱的耐受性,其热稳定性远逊于Lac/4-MBA@GNP/Au,催化活性随温度升高逐渐下降,当温度升至313 K时,催化活性就下降到了不到初始值的1/3,这可以归因于温度升高使载体表面物理吸附的酶分子脱附 (温度升高前后溶液中铜离子浓度有明显变化且溶液中加入ABTS会变成浅绿色,证明酶分子从电极表面脱落进入溶液)。
2.3 组装固酶燃料电池的性能
支持信息图S6为固酶阳极GOx/4-MBA@GNP/Au在含不同浓度葡萄糖的0.2 mol·L-1无氧PBS中以相同扫速扫描所得CV曲线,从图S6可以看出,当PBS中加入葡萄糖后,在-0.03 V附近氧化电流开始明显增加,在0.2 V左右达到极限同时阴极峰电流迅速降低,随着加入葡萄糖浓度的增加,极限催化氧化电流也随之上升,直至葡萄糖浓度足够高(>60 mmol·L-1),达到饱和催化电流为止(31μA左右),由此按照2.2.2节相似方法,根据前述给出的导电GOx分子表面浓度和电极活性表面积,可以估算出固酶催化底物葡萄糖氧化的速率为1.3 s-1,相对于固定Lac催化氧分子转化速率0.5 s-1而言处于一个数量级但却是后者数值的2.6倍,有鉴于此,当将2个固酶电极组装为燃料电池时,不需要在两极室之间以常用的NAFION离子交换膜分隔开,因此降低了输出能量损失。需要指出的是GOx/Py-GNP/Au既不能实现酶与纳米金粒子之间的直接电子迁移,也观察不到催化葡萄糖氧化的电信号,只能如文献[26]报道的体系那样,在引入外加电子中介体的情况下才可以实现有效催化葡萄糖氧化,测试结果类似文献[26]。
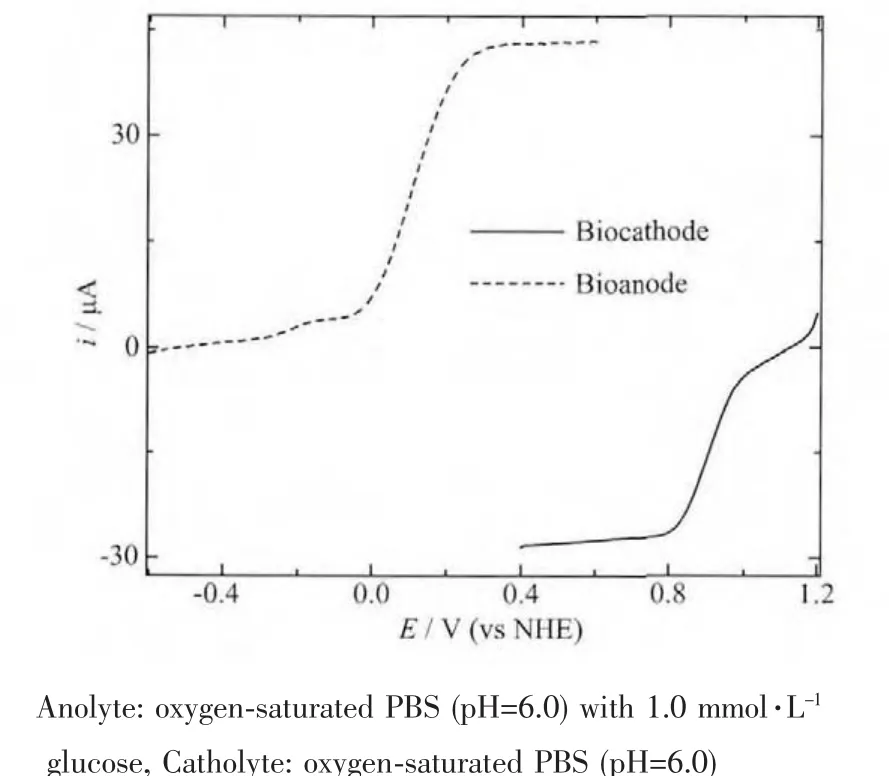
图9 固酶阳极GOx/4-MBA@GNP/Au和固酶阴极Lac/4-MBA@GNP/Au分别在阳极和阴极电解液中以100 mV·s-1扫描所得的极化曲线Fig.9 Polarization curves of bioanode GOx/4-MBA@GNP/Au and biocathode Lac/4-MBA@GNP/Au recorded in anolyte and catholyte at scan rate:100 mV·s-1
图9 为固酶阳极GOx/4-MBA@GNP/Au在含葡萄糖(1 mmol·L-1)的氧气饱和 PBS(pH=6.0)中,固酶阴极Lac/4-MBA@GNP/Au 在氧气饱和 PBS(pH=6.0)中,以相同扫速获得的极化曲线(固酶自组装层数都是5),从图9可看出固酶阳极氧化电位始于-0.03 V,阴极还原起始电位在0.96 V附近,当反应为底物扩散控制时,催化电流达到极限。值得注意的是,当阴阳极底物浓度相近时,阳极极限催化电流是阴极极限催化电流的近2倍,这表明阴阳极反应均受底物扩散控制而且此电池的能量输出受限于阴极氧还原过程,这与前述讨论得到结论相一致。阴极还原过程成为决速步的原因可能源于固酶载体表面的固酶纳米金粒子团簇过于庞大,不利于氧气传质,加上酶与氧分子有效接触不充分,降低了酶的有效催化活性,当固酶层数增加之后,此现象会更为显著(与2.2.2节结论相一致)。
由GOx/4-MBA@GNP/Au和Lac/4-MBA@GNP/Au组成4-MBA@GNP固酶基生物燃料电池 (无NAFION隔膜,电解液为含葡萄糖 1.0 mmol·L-1的 O2饱和PBS,pH=6.0),其极化和性能曲线见图10。由GOx/Py-GNP/Au和Lac/Py-GNP/Au组成Py-GNP/Au固酶基生物燃料电池 (阴阳极室以厚度0.125 mm为NAFION隔膜Nafion 115分隔,阳极电解液为含葡萄糖1.0 mmol·L-1+0.5 mmol·L-1二茂铁甲酸 (FMCA) 的无氧PBS,pH=6.0,阴极电解液为O2饱和的含ABTS 1.0 mmol·L-1PBS,pH=6.0), 其极化和性能曲线见支持信息图S7。从图 10和支持信息图 S7可以看出,4-MBA@GNP固酶基生物燃料电池的开路电压(OCV)约为0.88 V,在0.24 V具有最大输出能量密度,大约是864.0μW·cm-2。由于固酶阴极上酶-电极间电子迁移速率不够快以及氧分子与固定酶反应不充分等原因,OCV与理论最大输出电压0.99 V还存在110 mV的电压差,但相对于其他文献[2,24]报道的类似纳米体系固酶电池的OCV和最大输出能量密度要高出很多,即使是与贵金属中心离子作为活性位点的氧化还原聚合物电子中介体的固酶体系[3,22]相比,能量输出性能也处在同一数量级而且还无需使用昂贵的贵金属试剂和复杂的合成路线,具有成本低且制备简单的优势。相比较而言,Py-GNP/Au固酶基生物燃料电池的OCV(0.46 V)和最大输出能量密度(77.0μW·cm-2)都比前者低很多,因为使用电子中介体和隔膜降低了电池的最大输出电压,中介体扩散速率较慢加之酶与电子中介体化学反应速率受电极表面固酶聚集形态影响,输出能量密度不高,更重要的是,随着电极使用时间的延长,由于酶,电子中介体的变性或电子传递性能的下降,使用电子中介体的酶燃料电池的长期使用性能一般比直接电子迁移型酶燃料电池差(图11):前者储存3 d后输出功率就下降到最大值的2/3左右,这与前述结论相一致;相对而言,能够实现酶-电极间直接电子迁移的自组装多层酶燃料电池不但具有更高的能量输出功率,而且具有优异的长期使用性能,储存3周后输出功率仍可达到最佳能量输出的80%以上,这与该电极上固酶载体对酶具有良好的亲和力以及适当的酶-载体间相互作用有很大关联。

图10 4-MBA@GNP固酶基葡萄糖/O2燃料电池的极化和能量输出性能曲线Fig.10 Polarization curve and energy out-put performance curve of 4-MBA@GNPwith immobilized enzymebased glucose/O2 fuel cell

图11 两种功能化纳米金粒子固酶基葡萄糖/O2燃料电池能量输出的长期稳定性Fig.11 Long-term stability in energy out-put performance of two kinds of functionalized nano-gold particles with immobilized enzyme-based glucose/O2 fuel cell
3 结 论
本文采用2种功能化纳米金粒子:Py-GNP和4-MBA@GNP作为固酶载体和导电基体,以化学偶联或物理吸附的方式固定Lac和GOx分子,同时利用聚合物/酶-纳米金粒子相互之间的氢键作用构筑自组装多层固酶电极,以电化学方法研究这些电极的直接电子迁移性能和催化底物反应性能,在此基础上评估了由这些电极组装的酶基燃料电池的能量输出性能,得到如下结论: (1)4-MBA@GNP作为固酶载体构筑的固酶阳极和固酶阴极都可以实现酶活性中心与导电基体之间的直接电子迁移,而且对相应底物-葡萄糖和氧气具有优异的催化性能,同时具有较快的电子迁移速率。(2)可实现直接电子迁移的4-MBA@GNP基固酶电极不但具有良好的重现性和长期使用性,而且酸碱耐受性和热稳定性都优于Py-GNP/Au基固酶电极,前者还可通过自组装方式获得多层固酶电极,以获得更高的催化性能,但当固酶层数过多时,催化性能会逐渐降低。(3)4-MBA@GNP基固酶电极组装得到的酶燃料电池相对于Py-GNP/Au基固酶电极组装成的酶燃料电池而言具有以下优势:无需引入电子中介体和隔膜,具有更高的OCV和更大的能量输出密度,以及更优良的长期使用性能。但这种电池由于底物扩散受限和酶-底物接触反应不充分等原因,电池输出性能还有提升的空间。
Supporting information is availableat http://www.wjhxxb.cn
[1]Coman V,Vaz-Dominguez C,Ludwig R,et al.Phys.Chem.Chem.Phys.,2008,10(40):6093-6096
[2]Willner I,Yan Y M,Willner B,et al.Fuel Cells,2009,9(1):7-24
[3]Ivanov I,Vidakovic-Koch T,Sundmacher K.Energies,2010,3(4):803-846
[4]Stolarczyk K,Nazaruk E,Rogalski J,et al.Electrochem.Commun.,2007,9(1):115-118
[5]Wei W,Li P P,Li Y,et al.Electrochem.Commun.,2012,22:181-184
[6]Lesniewski A,Niedziolka-Jonsson J,Rizzi C,et al.Electrochem.Commun.,2010,12(1):83-85
[7]Arrocha A A,Cano-Castillo U,Aguila S A,et al.Biosens.Bioelectron.,2014,61:569-574
[8]Katz E,Riklin A,Heleg-Shabtai V,et al.Anal.Chim.Acta,1999,385(1/2/3):45-58
[9]Zelechowska K,Stolarczyk K,Lyp D,et al.Biocybern.Biomed.Eng.,2013,33:235-245
[10]Umasankar Y,Brooks D B,Brown B,et al.Adv.Energy Mater.,2014,4(6):1-9
[11]Kizling M,Stolarczyk K,Kiat J S S,et al.Electrochem.Commun.,2015,50:55-59
[12]Deng MF,Zhao H,Zhang SP,et al.J.Mol.Catal.B:Enzym.,2015,112:15-24
[13]Blandford CF,Heath R S,Armstrong F A.Chem.Commun.,2007,43:1710-1712
[14]Pang H L,Liu J,Hu D,et al.Electrochim.Acta,2010,55(22):6611-6616
[15]Qiu H J,Xu C X;Huang X R,et al.J.Phys.Chem.C,2008,112(38):14781-14785
[16]Lioubashevski O,Chegel V I,Patolsky F,et al.J.Am.Chem.Soc.,2004,126(22):7133-7143
[17]Pita M,Shleev S,Ruzgas T,et al.Electrochem.Commun.,2006,8(5):747-753
[18]Thorum M S,Anderson CA,Hatch JJ,et al.J.Phys.Chem.Lett.,2010,1(15):2251-2254
[19]Rahman M A,Noh H B,Shim Y B.Anal.Chem.,2008,80(21):8020-8027
[20]Zayats M,Katz E,Baron R,et al.J.Am.Chem.Soc.,2005,127(35):12400-12406
[21]Hao E C,Lian T Q.Chem.Mater.,2000,12(11):3392-3396
[22]Szamocki R,Flexer V,Levin L,et al.Electrochim.Acta,2009,54(7):1970-1977
[23]HUANG Jun(黄俊),ZHOU Ju-Ying(周菊英),XIAO Hai-Yan(肖海燕),et al.Acta Chim.Sinica(化学学报),2005,63(14):1343-1347
[24]Zhao H Y,Zhou H M,Zhang JX,et al.Biosens.Bioelectron.,2009,25(2):463-468
[25]Shleev S,Christenson A,Serezhenkov V,et al.Biochem.J.,2005,385:745-754
[26]Liu Y,Wang M K,Zhao F,et al.Biosens.Bioelectron.,2005,21(6):984-988
[27]Palmer A E,Randall D W,Xu F,et al.J.Am.Chem.Soc.,1999,121(30):7138-7149
[28]Dimcheva N,Horozova E.Biochemistry,2013,90:1-7
[29]Zeng H,Tang Z Q,Liao L W,et al.Chin.J.Chem.Phys.,2011,12(36):10888-10895
[30]Tsujimura S,Kamitaka Y,Kano K,et al.Fuel Cells,2007,7(6):463-469
[31]Stolarczyk K,Sepelowska M,Lyp D,etal.Bioelectrochemistry,2012,87:154-163
[32]Jiang D S,Long SY,Huang J,et al.Biochem.Eng.J.,2005,25(1):15-23
[33]Clot S,Gutierrez-Sanchez C,Shleev S,et al.Electrochem.Commun.,2012,18:37-40
[34]Mano N,Kim H H,Zhang Y C,et al.J.Am.Chem.Soc.,2002,124(22):6480-6486
[35]Liu Y,Qu X H,Guo H W,et al.Biosens.Bioelectron.,2006,21(12):2195-2201
猜你喜欢
化工管理(2022年14期)2022-12-02
有色设备(2022年2期)2022-08-06
化工管理(2021年7期)2021-05-13
陶瓷学报(2021年1期)2021-04-13
军民两用技术与产品(2021年10期)2021-03-16
中学生数理化(高中版.高二数学)(2020年2期)2020-04-21
商品与质量(2019年42期)2020-01-17
电子制作(2018年12期)2018-08-01
中国资源综合利用(2016年4期)2016-01-22
