水溶性纳米Fe3O4-阿魏酸抗凝血材料的合成及性能
2015-09-15 01:41:02高琦宽王喜存宋玉民
无机化学学报 2015年12期
高琦宽 王喜存 宋玉民*,
(1甘肃卫生职业学院,兰州 730000)
(2西北师范大学化学化工学院,兰州 730070)
近年来,血栓栓塞类疾病越来越受到人们的关注,它是一类严重危害人类健康和生命的疾病,可发生于全身各处血管中,从而表现出各种症状,是中风、心梗塞、脑梗塞的直接原因之一[1-3]。华法灵钠、肝素、枸橼酸钠是目前通用的抗凝血药物,但经常使用或大量使用非常容易引起自发性出血,所以研究和开发更好的抗凝血药物,成为药学专家和生物化学家的一个目标,如焦天权,朱元成等曾研究过稀土元素配合物的抗凝血性质[4-5]。从当归、川穹、阿魏、升麻中提取的阿魏酸(Ferulic acid,化学名称为4-羟基-3-甲氧基肉桂酸,Scheme 1),具有抗氧化、抗动脉粥样硬化、抗炎、抗凝血等多种药理学作用,因此广泛应用于药物、食品、保健等领域,临床上可用于心脏病、脑血管病、某些血液病等疾病的治疗[6-8]。但是阿魏酸(FA)分子中含有苯环结构,属于憎水基团,很难溶于水和透过生物膜双分子层,所以对阿魏酸分子进行改性以得到阿魏酸衍生物或制备水溶性阿魏酸金属配合物成为研究的热点之一[9]。本课题组曾研究过过渡金属与阿魏酸二元配合物、稀土元素与阿魏酸二元配合物的抗凝血作用[10-11],结果表明这些二元配合物的抗凝血时间比阿魏酸的抗凝血时间要长,但二元配合物的水溶性仍然较差。由于阿魏酸及其衍生物的水溶性差,直接影响到其药用效果,如何提高其药物的利用效率和靶向给药性,一直是药物学家和化学家关注的课题。Wang等通过化学修饰的方法将DBI-PEG水溶性长链分子接枝到Fe3O4磁性纳米粒子的表面[12],在水溶性长链的末端通过席夫碱(-C=N-)把抗癌药物色酮甲醛接枝到Fe3O4磁性纳米粒子上,药物的水溶性大大提高,而且由于Fe3O4磁性纳米粒子的定向作用,药物达到了更好的治疗效果。针对阿魏酸水溶性差的特性,作者参考Wang等的方法,先在DBI-PEG水溶性长链分子末端通过化学反应将阿魏酸分子接枝上去,后将已接枝阿魏酸分子的DBI-PEG水溶性长链分子接枝 到Fe3O4磁性纳米粒子的表面,得到了水溶性较好的Fe3O4-DBI-PEG-阿魏酸长链分子 (溶解度>10 mg·mL-1,而阿魏酸溶解度<1 mg·mL-1)。抗凝血实验表明,Fe3O4-DBI-PEG-阿魏酸长链分子,比阿魏酸分子的抗凝血时间、复钙时间要长。说明Fe3O4-DBI-PEG-阿魏酸比阿魏酸分子具有更好的抗凝血性质。本文报道了Fe3O4-DBI-PEG-阿魏酸磁性纳米粒子杂化材料的合成、表征及体外抗凝血性质研究结果。在外磁场存在下的该杂化材料在动物体内的靶向给药性研究正在进行中。
1 实验部分
1. 1 仪器和试剂
FTS-3000型红外光谱仪 (美国PE公司,KBr压片, 摄谱范围 4 000~400 cm-1);Mercury plus 400 超导核磁仪(美国Varian公司),TG/DTA-6300热重-差热分析仪(美国PE公司);数显智能控温磁力搅拌器(巩义市英峪予华仪器厂);恒温水浴锅(郑州长城科工贸有限公司);KQ-100M超声波清洗器 (东莞市科桥超声波设备有限公司)。产品的表面形貌的表征采用的仪器是扫描电子显微镜(XL-20,飞利浦公司,荷兰),操作电压为25 kV;透射电镜 (Philips EM420 120 kV);振动样品磁强计(美国 Lake shore 7404);激光粒度仪 (Zetasizer Nano-Zs laser particle size instrument,Malvern,UK)。 聚乙二醇 4000,CH2Cl2,三乙胺,CH2SO2Cl,3,4-二羟基苯甲醛均为分析纯。Fe3O4纳米粒子 (20 nm)购自Aladdin Chemistry Co.Ltd,阿魏酸(上海中秦化学试剂有限公司)。
1.2 杂化材料的合成
1.2.1 合成 H2N-PEG-NH2
在250 mL烧瓶中加入5 mmol聚乙二醇4000和150 mL干燥后的CH2Cl2,再加入15 mL三乙胺,混合冰浴下逐滴加入7 mL CH3SO2Cl。加完后移去冰浴,室温下反应 24 h,用 150 mL 50 mmol·L-1的NaHCO3溶液洗涤后用CH2Cl2萃取过滤。
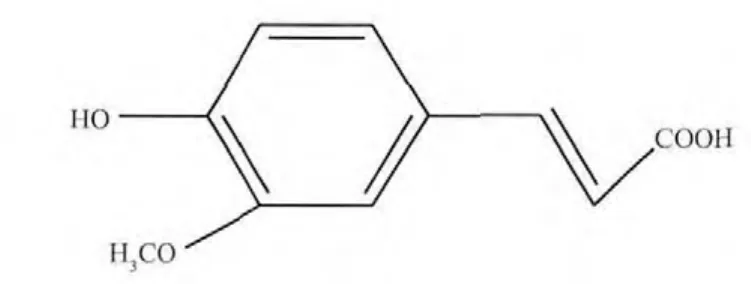
Scheme 1 Molecular structure of ferulic acid
1.2.2 合成 DBI-PEG-NH2
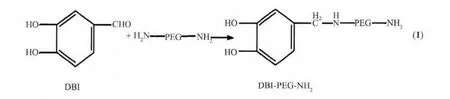
称取 0.2 mmol H2N-PEG-NH2,加 25 mL 乙醇和25 mL CH2Cl2于圆底烧瓶中, 称取0.2 mmol 3,4-二羟基苯甲醛,用50 mL乙醇溶解后置于恒压漏斗中,缓慢滴加进入圆底烧瓶(12 h),同时冰浴(反应过程如(1)式)。 再加入 0.2 mmol NaBH4搅拌 4 h。 常压过滤,蒸干后加乙醚抽滤,得到产物,产率40%[13]。
1.2.3 合成阿魏酸杂化材料
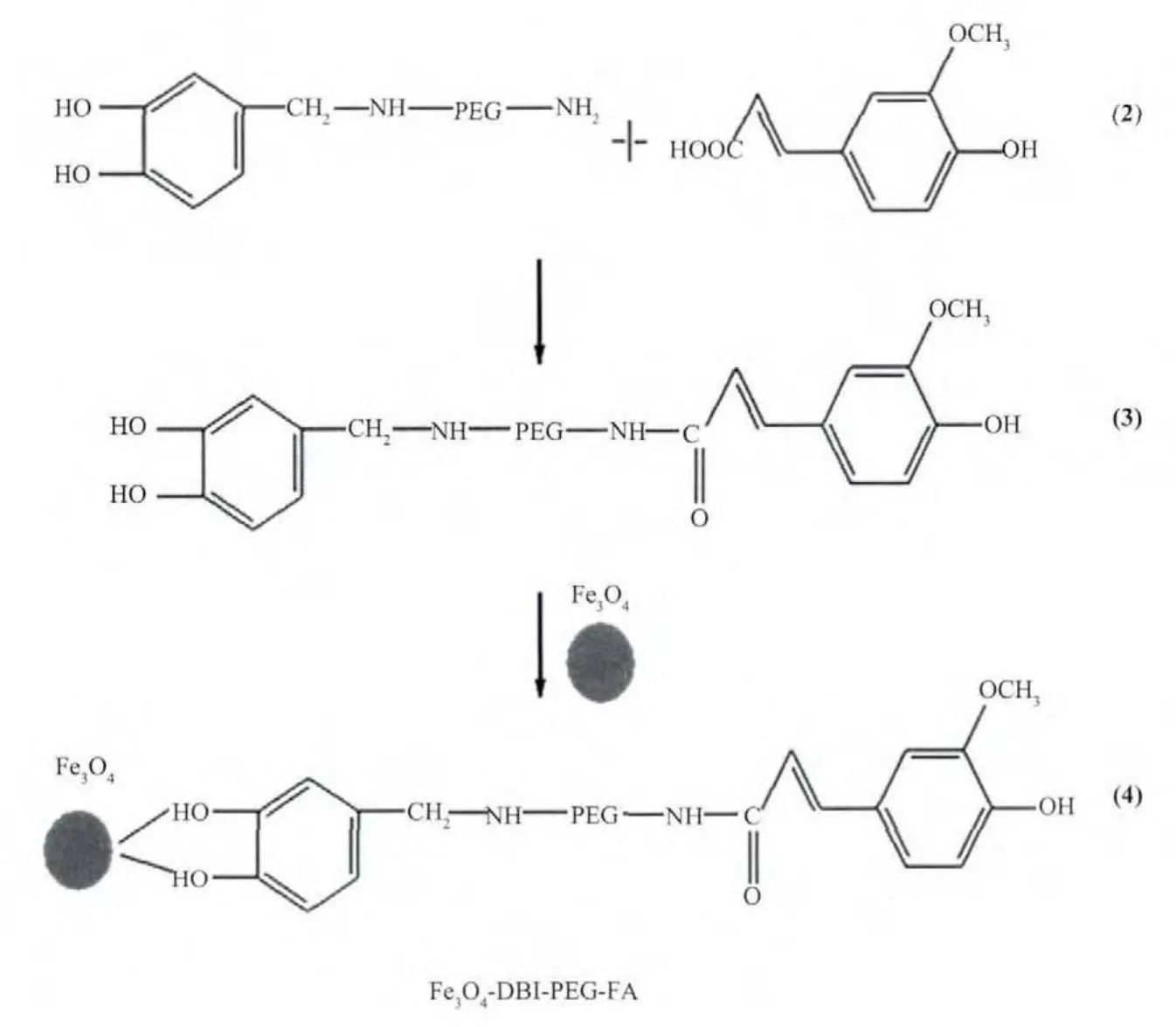
分别称取 0.25 mmol阿魏酸,0.03 mmol DCC(N,N′-二环己基碳二亚胺,脱水缩合剂)和0.05 mmol NHS(N-羟基丁二酰亚胺,酰胺化试剂)于圆底烧瓶中,用15 mL DMF进行溶解,在常温下搅拌 24 h后再加入 DBI-PEG-NH20.25 mmol,氯仿 3 mL,完全溶解后搅拌12 h(反应过程如(2)、(3式)),后加入氯仿分散的Fe3O4,无沉淀生成(反应过程如(4)式)。继续搅拌12 h后将溶液转移到离心管内加石油醚高速离心,分离离心管上层清液。将油状沉淀用二氯甲烷和石油醚处理成固体沉淀后再离心,去掉离心管上层清液。用乙醇清洗沉淀表面,晾干,得到粉末状产物,标记为Fe3O4-DBI-PEG-FA。
1.2.4 杂化材料溶液的配制
杂化材料用二次蒸馏水溶解,并超声10 min,配制成 2.0,0.20,0.020 mmol·L-1的溶液。
1.3 凝血实验
1.3.1 全血凝血实验(CT)
取3组干净的试管,每组3支。第1组为空白组,第2、3组分别为阿魏酸(FA)、阿魏酸杂化材料(加 入 溶 液 的 浓 度 均 为 2.0 mmol·L-1, 体 积 为0.25 mL)。
将空白组试管和加好样品的2组试管放入 37℃水浴中进行恒温,之后沿各试管壁加入1 mL人血,继续在水浴中恒温2 min,同时开启秒表,每隔30 s以30°轻轻倾斜试管并观察,直至血液不再流动为止,记录该时间为全血凝血时间。
运 用 上 述 实 验 方 法 测 定 0.20 mmol·L-1和0.020 mmol·L-1溶液的凝血时间。
1.3.2 复钙实验(RT)
将阿魏酸及Fe3O4-DBI-PEG-FA固体放入0.154 mol·L-1的生理盐水中浸泡 24 h,烘干。取 3 组洁净的试管,每组3支。第1组为空白组,第2、3组分别加入0.1 g的FA,FA杂化材料,37℃水浴恒温2 min。 向 3组 试管 中分 别加入 人 血 0.3 mL,0.025 mol·L-1的 CaCl2溶液 0.3 mL,置于 37 ℃水浴中恒温,同时开启秒表,将一根不锈钢小钩伸入溶液中均匀缓慢地搅动,并检查是否有纤维蛋白的形成。当小钩上刚出现丝状物时,记录时间,此时间即为复钙时间。
1.3.3 抗凝血活性实验
采用活化部分凝血活酶时间(APTT)和凝血酶原时间 (PT)2个指标来考查杂化材料对凝血系统的影响[13]。
取5组洁净的试管,每组3支,第1组为空白样, 第 2~5 组分别加入浓度为 0.20 mmol·L-1和0.20 mmol·L-1的阿魏酸及阿魏酸杂化材料溶液0.25 mL。 向 15支试管中分别加入 0.5 mL血浆,0.025 mol·L-1的 CaCl2溶液 0.3 mL,37 ℃水浴恒温后,记录出现纤维蛋白丝的时刻,该时间为APTT。凝血酶原时间实验中,快速加入PT试剂,记录凝固时间,该时间为PT。
2 结果与讨论
2.1 杂化材料表征
2.1.1 红外光谱
在H2N-PEG-NH2红外光谱图中,出现很强的-C-H 伸缩振动吸收峰(2 887 cm-1,),很强的 νa(-C-OC)振动吸收出现在1 111 cm-1,中等强度的(CH2)吸收出现在1 467 cm-1;在DBI-PEG-NH2的红外谱中,不仅出现有H2N-PEG-NH2的基团特征吸收峰(2 887、1 112 cm-1), 还 出 现 ν(-OH)的 吸 收 峰(3 445 cm-1),说明3,4-二羟基苯甲醛与H2N-PEGNH2反应,生成DBI-PEG。在Fe3O4-DBI-PEG红外光谱图中,不仅有DBI-PEG-NH2分子基团的特征吸收峰(3 445、2 887、1 112 cm-1),还出现很强的金属 Fe-O吸收峰(616 cm-1)。
在阿魏酸、Fe3O4-DBI-PEG-阿魏酸杂化材料的红外图谱中可以看出阿魏酸与杂化材料相比某些特征吸收峰发生了明显的位移,强度也有着相应的变化,说明阿魏酸接枝到纳米氧化物表面。阿魏酸分子羧酸中O-H的面外变形振动吸收峰出现在950~900 cm-1[14],羧羰基振动峰出现在1 686 cm-1,在3 425 cm-1处的酚羟基伸缩振动吸收峰,形成杂化材料后变成了一个平滑的宽峰 (3 300~3 600 cm-1),是阿魏酸杂化材料中O=C-N基团中的胺基 (3 300~3 500 cm-1)振动吸收峰与阿魏酸酚羟基的伸缩振动吸收峰的叠加结果。阿魏酸在1 686 cm-1处的羰基振动峰在杂化材料的红外谱图出现在1 635 cm-1处,向低波数移动,说明阿魏酸的羧羟基与PEG的NH2发生作用,生成了酰胺基团。由此可见阿魏酸杂化材料被成功合成。
2.1.2 杂化材料的1H NMR分析
阿魏酸在 CDCl3中1H NMR 为:δ=3.83(-OCH3),6.37(-CH=C-benzene),7.54 (-benzene-CH=C),9.57(-OH),12.16(-COOH)。H2N-PEG-NH2在 CDCl3中1H NMR 为:δ=2.49 (-NH2),3.33 (N-CH2),3.49(-CH2)。DBI-PEG-NH2在 CDCl3中1H NMR 为 : δ=2.97(t,2H,NH2CH2),3.63(brs,ca.373H,PEG 4000),3.68(t,2H,NH2CH2CH2),6.45(d,1H,Ph),6.44(d,1H,Ph),6.36 (s,1H,Ph)。 DBI-PEG-FA 在 CDCl3中1H NMR 为:δ=2.32(-NH),6.45(-CH=C-benzene),7.56(-benzene-CH=C),3.79(-OCH3),10.51(-OH)。
显然形成杂化材料以后,-NH中的1H NMR由最初的 2.49 位移至 2.32,-CH=C-benzene 由 6.37 位移 到 6.45,-benzene-CH=C 由 7.54 位 移 到 7.56,-OCH3由 3.83 位 移 到 3.79,-OH 由 9.57 位 移 到10.51,而-COOH的1H NMR消失,说明阿魏酸已接枝到H2N-PEG-NH2的一端,与DBI和Fe3O4共同形成了杂化材料。
2.1.3 杂化材料的热分析
以α-Al2O3为参比,以10℃·min-1为升温速率,在N2保护下从室温升至800℃,得阿魏酸和杂化材料的热重(TG-DTA)曲线。图1为阿魏酸(a)和阿魏酸杂化材料(b)的TG曲线图。
从图1中可以看出,对于Fe3O4-DBI-PEG-NH2的热重曲线(a),在25~220℃温度范围内失重5%左右,是该物质失去的自然吸附水造成的。第二阶段在230~450℃温度范围内,失重 26.0%,这是失去的嫁接在纳米四氧化三铁表面的DBI-PEG-NH2所致。然而对于Fe3O4-DBI-PEG-FA的热重曲线(b),在第一阶段25~250℃失重2.0%的自然吸附水,而在250~650℃急剧失重约58.0%,这部分是因为失去了嫁接在Fe3O4纳米粒子表面的DBI-PEG-FA,最终产物为Fe3O4。接枝的阿魏酸大约占3.0%,所有这些信息说明Fe3O4-DBI-PEG-阿魏酸分子被合成了。
与文献报道的阿魏酸分解温度190℃相比[15],杂化材料的分解温度在250℃以上,表明杂化材料的稳定性大于阿魏酸。
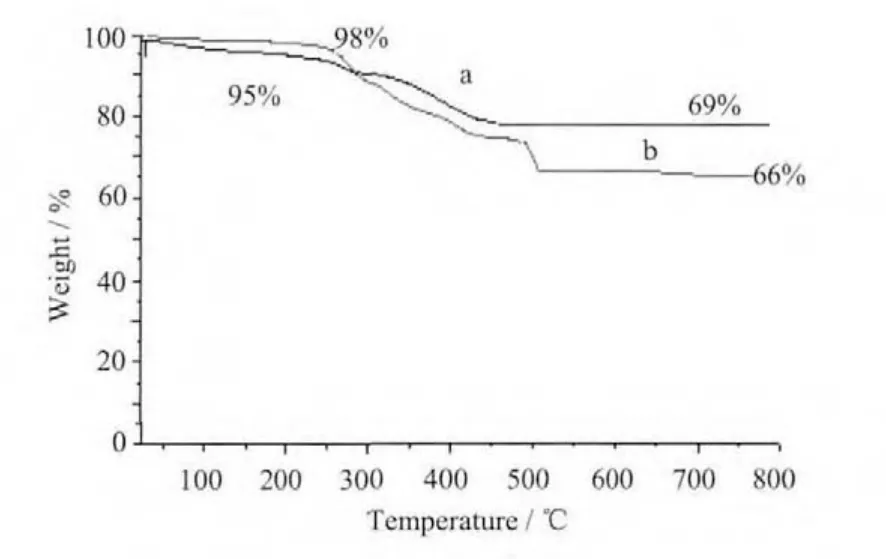
图1 (a)Fe3O4-DBI-PEG-NH2和(b)Fe3O4-DBI-PEGFA热重图Fig.1 TG of Fe3O4-DBI-PEG-NH2(a)and Fe3O4-DBIPEG-FA(b)
2.1.4 合成材料的电镜图分析

图2 Fe3O4和Fe3O4-DBI-PEG-FA的电镜图Fig.2 SEM images of Fe3O4 nanoparticles(a)and Fe3O4-DBI-PEG-FA(b);TEM images of Fe3O4 nanoparticles(c)and Fe3O4-DBI-PEG-FA(d)
图2 为Fe3O4和Fe3O4-DBI-PEG-FA的电镜图。从电镜图可以看出有一定团聚现象的球形纳米Fe3O4粒子,如图2a。当纳米Fe3O4粒子通过DBIPEG-NH2嫁接到阿魏酸上后,由于分子体积变大导致粒径增大,粒子更加分散,长度在40~100 nm不等,杂化材料分别为层状和纳米纤维状结构的固体,进一步说明纳米氧化物被DBI-PEG-NH2改性,并和阿魏酸反应接枝,在扫描电镜中基本看不到纳米氧化物粒子,这表明在纳米氧化物表面成功的发生了接枝反应,阿魏酸已将氧化物粒子完全包覆,如图2b。但在透射电镜图中(图2c),可以明显看出球形聚集的纳米Fe3O4粒子,还可以看出单独的纳米Fe3O4粒子和在阿魏酸杂化材料(图2d)中的纳米粒子非常相似,由于透射电镜无法反映有机基团,只能看到纳米粒子周围存在有白色物质,说明Fe3O4粒子被有机物DBI-PEG-FA包裹。
2.1.5 合成材料的磁性能分析
由图3可以看到在相同强度磁化场内,Fe3O4纳米粒子单位质量的磁感强度较大而Fe3O4-DBI-PEGFA粒子单位质量的磁感强度较小。这说明两种物质都含有Fe3O4磁性粒子。Fe3O4纳米粒子磁感强度较大是因为样品全部是Fe3O4粒子,而Fe3O4-DBI-PEGFA粒子的磁感强度较小是因为样品含有部分Fe3O4粒子部分有机基团,导致磁性的Fe3O4粒子的含量降低。由此可说明有机基团的引入导致Fe3O4-DBIPEG-FA单位质量的磁感强度降低。
由于引入Fe3O4纳米粒子,杂化材料表现出顺磁性。合成的杂化材料的水溶液在没有外磁场的情况下能够稳定存在(图4a)。当有外磁场存在时,该溶液中的杂化材料分子就会向磁场方向移动,聚集到一起,如图4b所示。杂化材料的这种顺磁性是该材料在动物体内可以具有靶向给药性的依据。
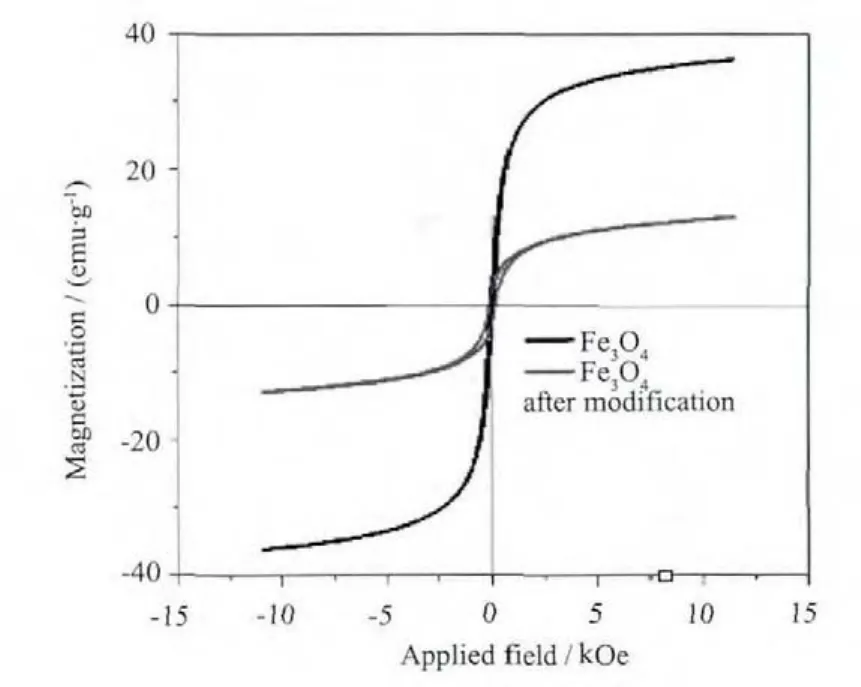
图3 纳米Fe3O4和Fe3O4-DBI-PEG-FA的振动样品磁强计图Fig.3 VSM images of Fe3O4 NPs and Fe3O4-DBI-PEGFA NPs
2.1.6 合成材料的粒径测试
图5是DBI-PEG和杂化材料Fe3O4-DBI-PEGFA的粒径分布强度,可以看出DBI-PEG的粒径大小约为为114 nm,而当其与阿魏酸接枝形成Fe3O4-DBI-PEG-FA后其粒径增大,为176 nm,说明阿魏酸已经接枝到了DBI-PEG上,形成了杂化材料。且其分布峰变宽,所形成的杂化材料表现出一定的团聚性。
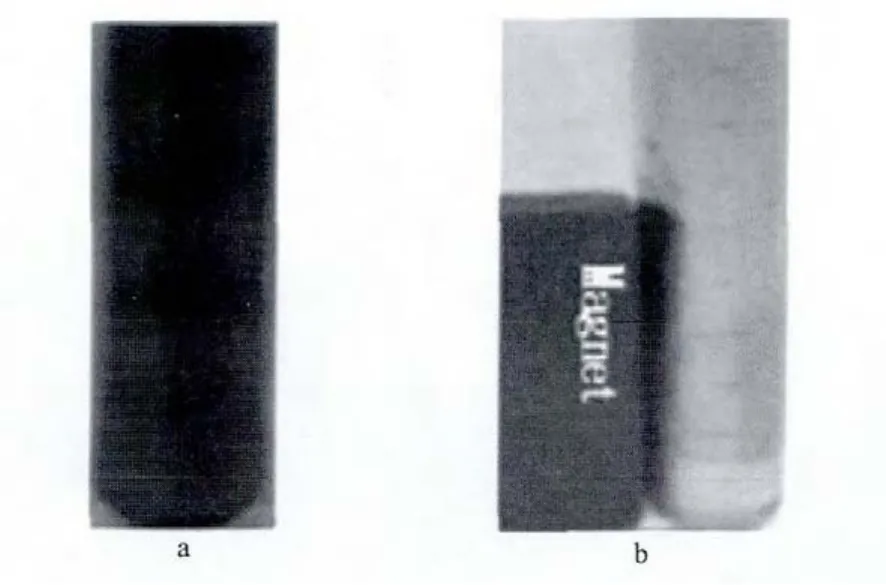
图4 杂化材料的磁性Fig.4 Hybrid materials solution without magnet(a)and with magnet(b)
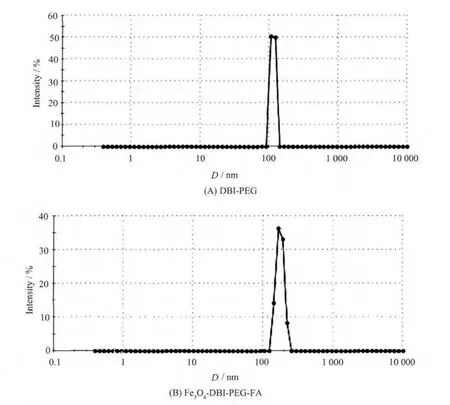
图5 粒径分布图Fig.5 Particle size distribution
2.2 抗凝血试验
2.2.1 全血凝血实验(CT)
表1为不同浓度的阿魏酸及杂化材料的凝血时间。从表中可以看出,空白样的凝血时间最短,其次是阿魏酸,相同浓度杂化材料的凝血时间均大于阿魏酸。从表中还可以看出,随着浓度的增加,阿魏酸和杂化材料的凝血时间均有延长。但是如果浓度过大,分子会在人体内大量累积,不利于新陈代谢,甚至会危害健康。在同一浓度时,与阿魏酸相比,杂化材料的凝血时间几乎是阿魏酸的2倍,这说明阿魏酸杂化材料具有较好的抗凝血性质。本实验合成的阿魏酸杂化材料加入血液后,影响凝血过程中凝血因子的激活及凝血复合物的形成[16],阻止了凝血酶原转化为凝血酶,因此,杂化材料的有效抗凝血时间得以延长。
2.2.2 复钙实验(RT)
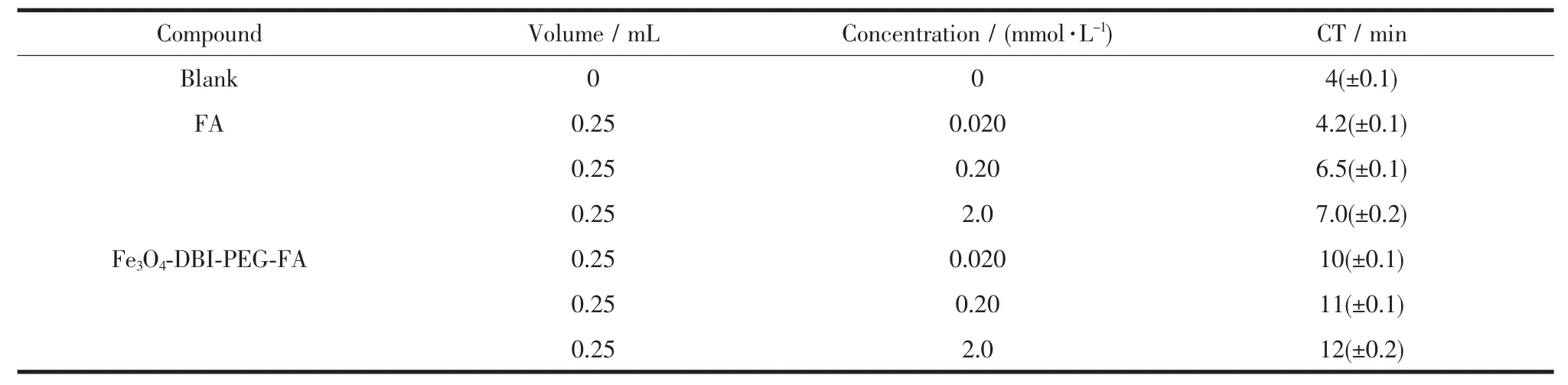
表1 不同浓度的Fe3O4-DBI-PEG-FA的凝血时间Table 1 Coagulation time of different concentrations compound
表2为阿魏酸杂化材料的复钙时间(RT)。从表中数据可知,不加任何物质的空白样的复钙时间最短,其次是阿魏酸,杂化材料复钙时间最长。因此,杂化材料的抗凝血性优于阿魏酸。与前面所做的凝血时间实验结果相一致。
2.2.3 凝血活性综合实验
活化部分凝血活酶时间(APTT)和凝血酶原时间(PT)是内源性凝血活性和外源性凝血活性的综合性指标。二者的延长表明存在先天性凝血因子异常或多种凝血因子的缺乏及循环抗凝血素的增加的可能性[13]。从表3的实验结果可以看出:(1)与空白组相比,阿魏酸和杂化材料的APTT、PT时间均有所延长,且APTT时间延长较多,这是由于在测试组的血液样品中加入了阿魏酸和杂化材料,说明不仅阿魏酸具有抗凝血作用,合成的杂化材料也具有抗凝血作用。(2)同等实验条件下,浓度较大的阿魏酸和杂化材料的溶液比浓度较小的阿魏酸和杂化材料溶液的APTT、PT时间稍有延长,这表明抗凝作用与测试样品的浓度有关,样品浓度越大,抗凝血时间越长。(3)在表中所列的实验条件下,阿魏酸和杂化材料的APTT、PT测试时间结果均在正常值的范围内,因此可以看出,当在体外血液中加入少量杂化材料时,没有引起血液性质的较大变化,此种杂化材料有望进入临床抗凝血药物的筛选范围。

表2 Fe3O4-DBI-PEG-FA的复钙时间Table 2 Recalcification time of Compound
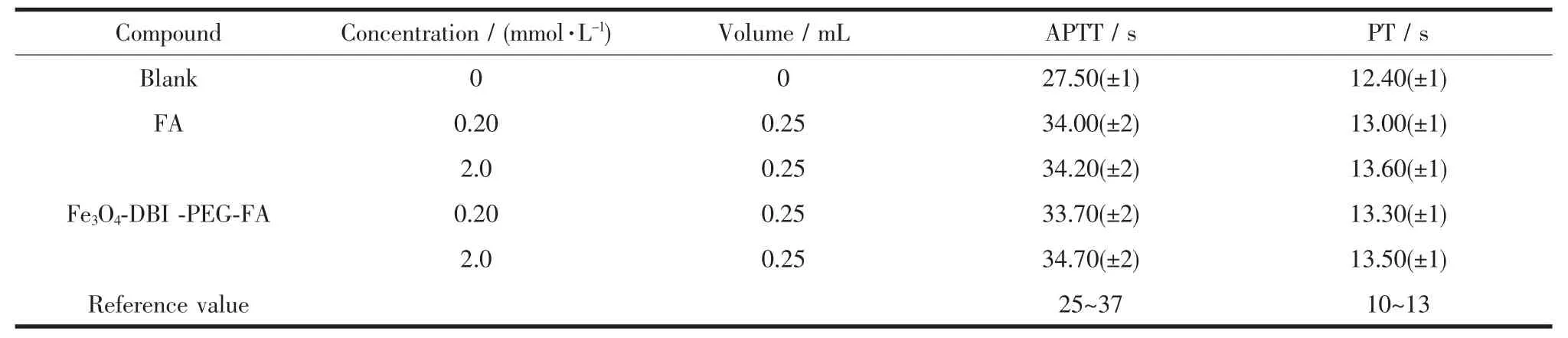
表3 Fe3O4-DBI-PEG-FA凝血活性实验Table 3 Coagulant activity of Compound
3 结 论
利用DBI-PEG4000活化纳米Fe3O4和接枝的方法制备了一类纳米氧化物阿魏酸杂化抗凝血材料,通过红外光谱、热重-差热分析和扫描电镜、透射电镜、磁强计表征产物,结果表明阿魏酸接枝在了经过DBI-PEG-NH2活化后的纳米Fe3O4氧化物表面。抗凝血实验说明杂化材料具有良好的水溶性和抗凝血性能。克服了阿魏酸水溶性差的缺点,相同条件下抗凝血时间和复钙时间比阿魏酸要长。杂化材料的抗凝血时间长短与材料的溶液浓度有直接的联系,浓度越大,抗凝时间越长。少量杂化材料在体外血液中的加入并没有引起血液性质的较大变化,此种杂化材料的抗凝血性能可为临床抗凝血药物的筛选提供实验依据。
[1]Yang M L,Song Y M.RSCAdv.,2015,5:17824-17833
[2]Eitsuka T,Tatewaki N,Nishida H,et al.Biochem.Biophys.Res.Commun.,2014,453(3):606-611
[3]Woranuch S,Yoksan R.Carbohydr.Polym.,2013,96(2):495-502
[4]Jiao T Q,Wu JG,Zeng F L,et al.Synth.React.Inorg.Met.-Org.Chem.,1999,29(5):725-735
[5]ZHU Yuan-Chen(朱元成),YAO Ka-Lin(姚卡玲),LI Yin(李英),et al.J.Lanzhou Univ.(兰州大学学报),2005,41(6):61-64
[6]YIN Hua-Fang(殷华芳),QIAN Xiao-Ping(钱晓萍),LIU Bao-Rui(刘宝瑞).Mod.J.Integr.Trad.Chin.West.Med.(现代中西医结合杂志),2010,19(32):4238-4240
[7]ZHAO Dong-Ping(赵东平),YANG Wen-Yu(杨文钰),CHEN Xin-Fu(陈兴福).Lishizhen Med.Mater.Medic.Res.(时珍国医国药),2008,19(8):1839-1841
[8]Zhao Z H,Mo H,Dai SA.Food Chem.,2008,109:691-702
[9]MA Fen-Shi(马逢时),LI Jia-Ming(李家明).Asia-Pac.Med.Trad.(亚太传统医药),2008,4(5):55-57
[10]WANG Pu-Yu(王璞玉),BIAN Chang-Xing(卞常鑫),SONG Yu-Ming(宋玉民).Chinese J.Inorg.Chem.(无机化学学报),2012,28(8):1609-1616
[11]SONG Yu-Ming(宋玉民),BIAN Chang-Xing(卞常鑫),WANG Pu-Yu(王璞玉).Chem.Bioeng.(化学与生物工程),2011,28(12):32-34
[12]Wang B D,Xu C J,Xie J,et al.J.Am.Chem.Soc.,2008,130(44):14437-14437
[13]CHEN Yu(陈于),XU Can(徐灿),SU Jia-Can(苏佳灿),et al.Chinese J.Inorg.Chem.(无机化学学报),2011,27(4):625-630
[14]ZHOU Hong-Hao(周宏灏).Pharmacology(药理学).Beijing:Science Publishing House,2007:318-329
[15]ZHOU Cai-Rong(周彩荣),AN Na(安娜),SHI Xiao-Hua(石晓华),et al.J.Zhengzhou Univ.(郑州大学学报),2007,28(2):47-48
[16]FANXiao-Li(樊小力).Physiology(生理学).Beijing:People′s Medical Publishing House,2002:54-60
猜你喜欢
合成树脂及塑料(2020年6期)2020-12-29 07:02:02
石油沥青(2019年4期)2019-09-02 01:41:54
陕西中医(2018年6期)2018-08-29 00:43:34
中国塑料(2016年3期)2016-06-15 20:30:03
中国塑料(2016年1期)2016-05-17 06:13:00
读写算·教研版(2016年8期)2016-05-07 11:52:08
中国塑料(2016年11期)2016-04-16 05:25:55
灾害医学与救援(电子版)(2016年3期)2016-03-11 20:18:06
中国卫生标准管理(2015年3期)2016-01-14 03:41:43
中国塑料(2015年1期)2015-10-14 00:58:41