ITO/g-C3N4异质结催化剂的制备及其光解水产氢性能
2015-09-15 01:41:00赵学国黄丽群李家科
无机化学学报 2015年12期
赵学国 黄丽群 李家科
(景德镇陶瓷学院,景德镇 333001)
近年来,随着光催化技术的发展,借助太阳能实现光催化技术在环境污染净化和太阳能转化为化学能领域方面表现了巨大的应用潜力。g-C3N4作为一种新型重要n型半导体材料,具有良好的化学和热稳定性,且成本低廉、易于制取,受到研究人员的广泛关注[1-2],但是其高的电子空穴复合率,较窄的可见光响应范围,低的电子传输效率等缺点使其应用受到严重的制约[3]。如何解决g-C3N4光催化材料的这些缺点成为研究人员思考的问题。采用非金属元素,如硫[4]、磷[5],和金属元素,如锌[6]、铁[7]等对 g-C3N4进行掺杂以提高其光催化活性已有报道。
半导体复合是近年来光催化剂材料研究的一个新方向,它将两种不同性质的导体或半导体材料组合在一起,使载流子作定向传输,可有效分离电子-空穴,提高了催化剂量子效率[8-9]。将g-C3N4与一些氧化物半导体,如 TiO2[10]、ZnO[11]等,或纳米碳材料,如碳纳米管[12]、石墨烯[13]等复合,可提高其光催化性能,这已为实验所证实。为提高g-C3N4催化剂光解水析氢的能力,本文采用热分解法先合成出g-C3N4粉体,通过静电引力作用将少量纳米ITO粒子修饰于其表面,有效地提高了其光生电子空穴的分离效率,并在可见光模拟系统中研究了附载的ITO对g-C3N4催化剂光解水析氢性能的影响。
1 实验部分
1.1 样品的制备
实验所用试剂为三聚氰胺(AR)、硝酸铟(AR)、四氯化锡(AR)、柠檬酸(AR),实验用水为去离子水。
1.1.1 氧化锡铟纳米粉的制备
1.1.2 g-C3N4催化剂粉体的制备
将一定量的三聚氰胺置于氧化铝坩埚中,用氧化铝坩埚盖盖上坩埚后移至马弗炉中于550℃反应3 h,并将升温速率控制在3℃·min-1,待其自然冷却后可制得g-C3N4粉体。
1.1.3 ITO/g-C3N4复合催化剂的制备
将一定量的氧化锡铟纳米粉加入到50 mL水溶液中,超声分散后待用。将一定量的g-C3N4催化剂粉体加入到300 mL水溶液中,超声分散后加入上述已分散的氧化锡铟纳米粉悬浊液,继续超声分散30 min,过滤并用清水反复清洗复合粉体后移入电热烘箱中于100℃烘干,制得浅黄色ITO/g-C3N4粉体。
1.2 样品表征
样品的结构表征采用日本理学DMAX-RB X射线粉末衍射仪 (XRD)完成,Cu Kα辐射,靶电压40 kV,电流 40 mA,λ=0.154 18 nm,石墨单色器滤波,扫描范围(2θ)10°~70°,扫描速度 0.02°·s-1。 粉末样品压片后进行衍射分析,采用闪烁计数器控测衍射线强度。采用日本JEM-2010型透射电子显微镜(TEM)研究样品形貌,阴极 LaB6,加速电压200 kV。采用Nicolet 7000-C型傅立叶红外光谱仪(波数4 000~400 cm-1,分辨率4 cm-1)对样品进行结构分析,试样采用高纯KBr与粉末样品压片进行红外光谱测试。采用Malvernζ电位测试仪在室温下于纯水中测试样品表面电位。禁带宽度测试采用Lamda 850紫外-可见近红外分光光度计。采用气相色谱仪(GC-950上海海欣色谱仪器有限公司)进行催化剂活性测试。
1.3 样品催化活性表征
光催化剂表面采用光还原原位负载1%(质量分数)纳米铂 (Pt)。将1 g光催化剂先分散于100 mL 20%(体积分数)乙醇水溶液中,向混合液中加入0.01 g氯铂酸铵,然后采用250 W氙灯进行照射30 min,过滤并烘干制得负载Pt的光催化剂。使用内置型光源(250 W氙灯)光反应器,气体闭合回路系统(氮气做载气),采用1 g催化剂,反应液为600 mL醇水混合液(V乙醇∶V水=1∶4),磁力搅拌,采用气相色谱对析氢量进行检测。
2 结果与讨论
2.1 物相结构与显微形貌分析
图1为实验中所制备的纯g-C3N4(曲线A)、1%ITO/g-C3N4(曲线 B)、2%ITO/g-C3N4(曲线 C)、3%ITO/g-C3N4(曲线 D)、4%ITO/g-C3N4(曲线 E)及 ITO(曲线F)的XRD图。
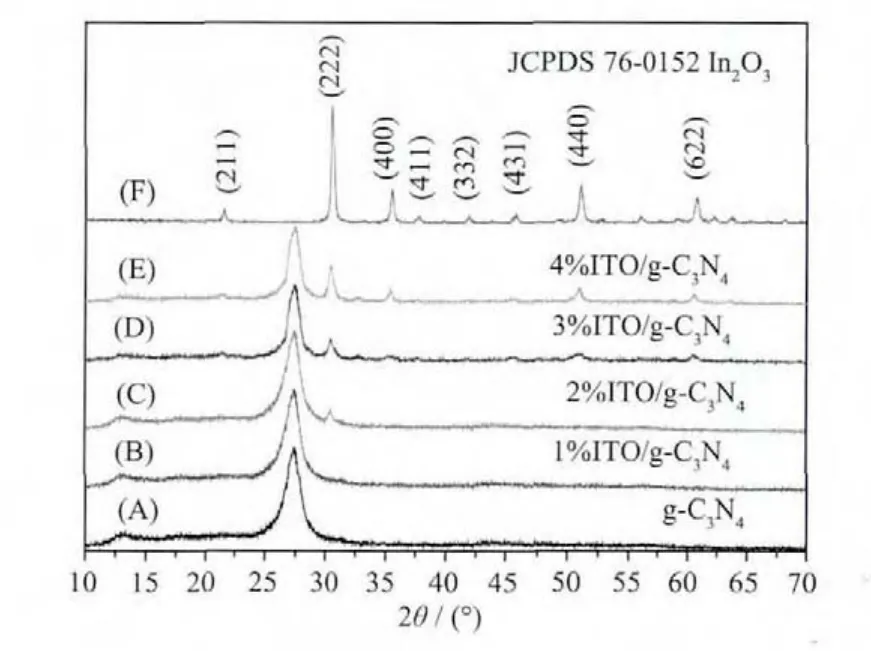
图 1 纯 g-C3N4(曲线 A)和 1%ITO/g-C3N4(曲线 B)、2%ITO/g-C3N4(曲线 C)、3%ITO/g-C3N4(曲线 D)、4%ITO/g-C3N4(曲线 E)和 ITO(曲线 F)的 XRD 图Fig.1 XRD patterns of pure g-C3N4(curve A)and 1%ITO/g-C3N4(curve B)、2%ITO/g-C3N4(curve C)、3%ITO/g-C3N4(curve D)、4%ITO/g-C3N4(curve E)and ITO(curve F)
从图1中可知,三聚氰胺经550℃热处理2 h后形成的氮化碳,其在 2θ=13.02°,27.4°处出现 2 个衍射峰,对应于石墨相C3N4(JCPDSNo.87-1526)的(100)和(002)晶面衍射峰[14];而经 550℃热处理 2 h后形成的氧化锡铟,其主要衍射峰与标准卡片(JCPDSNo.76-0152) 中立方相 In2O3的 (211)、(222)、(400)、(411)、(332)、(431)、(440)和(622)晶面衍射峰相一致。同时,从该图中还可以观察到,样品中引入的ITO对g-C3N4衍射峰峰位和强度未产生影响,这可能是其引入量较少及其复合方式的缘故。比较曲线C、D、E中各衍射峰可知,随着ITO在g-C3N4粉体中复合量的增多,其在(222)晶面衍射峰相对强度也在逐步增强。
图2A、2B为热分解法所制备g-C3N4粉体SEM显微照片及其相应微区能谱图。从图2A中可观察到,由低温热分解法所合成的g-C3N4粉体颗粒呈片状结构,宽约 1~2 μm,长约 2~10μm,厚约 100 nm,BET测试表明其比表面积约为10 m2·g-1。对图2A微区成份分析结果如图2B所示,各元素原子百分比为:C:42.45%,N:57.55%,故 nC/nN=0.74,与 g-C3N4中nC/nN配比非常接近。

图2 低温热分解法所制备g-C3N4粉体SEM显微照片(A)及其微区能谱图(B)Fig.2 SEM image of g-C3N4 powder(A)by thermal calcination and its corresponding EDS(B)
图 3A、3B、3C分别为 3%ITO/g-C3N4粉体 TEM显微照片、图3A方形区颗粒边缘高倍成像图及图3B方形区颗粒边缘高分辨成像图。图3A进一步清晰表明,热分解法所合成的g-C3N4粉体颗粒呈片状结构,且在颗粒表面及边缘处分布着一些异相纳米粒子。为了观察这些异相纳米粒子,分别对图3A和图3B中方形区颗粒边缘进行高倍成像。对单个颗粒边缘进行高倍成像(图3B)后可观察到,C3N4颗粒表面边缘有一些粒径在10~20 nm异相颗粒。单个异相粒子高分辨TEM图像如图3C所示,从图中可知,其晶面间距为0.292 nm,对应于立方相In2O3(222)晶面,说明,热分解法所合成的ITO纳米粒子可分散在g-C3N4颗粒子表面上。

图3 (A)为3%ITO/g-C3N4催化剂粉体TEM显微照片,(B)(C)分别为图3A、3B中方形区HRTEM照片Fig.3 (A)TEM image of 3%ITO/g-C3N4 catalyst,(B)the HRTEM image of the local enlarged image from the boxed region in Fig.3A,and(C)the HRTEM image of the local enlarged image from the boxed region in Fig.3B
2.2 ζ-电位分析
在实验过程中,通过简单的超声分散技术就能将ITO纳米粒子分散在g-C3N4颗粒表面形成异质结结构,其原因值得探讨。为此,我们对两种粉体进行了ζ电位分析。实验中所制备的g-C3N4和ITO颗粒在纯水(pH=7)中ζ-电位如图4所示。从图中可见,在纯水中 g-C3N4颗粒表面带负电 (ζ=-25 mV),而ITO颗粒带正电(ζ=19 mV),二者电性相反,在静电力作用下可形成异质结结构。

图4 g-C3N4和ITO颗粒在纯水(pH=7)中ζ-电位图Fig.4 ζ-potential of g-C3N4 and ITOpowder in water(pH=7)
2.3 傅立叶红外光谱及紫外可见光漫反射吸收光谱分析

图 5 g-C3N4(曲线 1)和 3%ITO/g-C3N4(曲线 2)粉体傅立叶红外光谱图Fig.5 FTIR spectrum of g-C3N4(curve 1)and 3%ITO/g-C3N4(curve 2)powder
g-C3N4(曲线 1)和 3%ITO/g-C3N4(曲线 2)粉体傅立叶红外光谱图如图5所示,曲线1中位于890、1 240 cm-1处的吸收峰为C-N键的伸缩振动吸收峰,1 640 cm-1吸收峰为C=N的振动模式,805 cm-1处的吸收峰可以归属为C-N-C基团的弯曲振动,3 130和3 355 cm-1吸收峰为N-H或O-H的伸缩振动峰[15]。比较曲线1和2可观察到,g-C3N4粉体与ITO复合后,在500 cm-1处出现了一较弱的吸收峰,为In2O3的特征吸收峰[16];在3 300 cm-1处出现了明显的强吸收峰,其原因可能是ITO与g-C3N4在水溶液中复合后致使复合粉体中含有较多的水份。
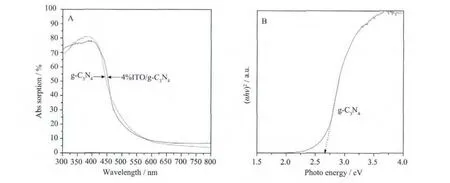
图6 x%ITO/g-C3N4(x=0,4)粉体紫外可见光吸收光谱(A)及其 (αhν)2-hν 曲线(B)Fig.6 UV-Vis absorption spectrum of x%ITO/g-C3N4(x=0,4)powder(A)and the(αhν)2 versus hνplot for determining the absorption onset of g-C3N4(B)
x%ITO/g-C3N4(x=0,4)粉体紫外可见光吸收光谱图如图6所示。从图6A中可以看出:(1)纯g-C3N4对可见光的吸收主要集中在300~500 nm之间;(2)当g-C3N4颗粒表面附着少量的ITO后,催化剂粉体对可见光的吸收未出现明显的变化,这主要是由于氧化铟较宽的带隙所致。此外,根据半导体的吸收系数α 与光学带隙之间的关系式(αhν)2=A(hν-Eg)作图(图6B所示)(α为吸收系数;Eg为禁带宽度),由g-C3N4可见光吸收光谱的数据可计算出g-C3N4禁带宽度Eg为 2.7 eV,这与文献所报道的结果相同[15]。
2.4 室温荧光光谱分析
图7为x%ITO/g-C3N4(x=0,2,3,4)的室温荧光光谱图,其激发光为200 nm的紫外光。从该图中可知,纯g-C3N4在420~575 nm之间存在强烈的荧光发射现象,其价带和导带带尾之间激子跃迁导致发光现象的产生。但是随着ITO纳米粒子对g-C3N4颗粒表面的修饰,其荧光强度及波长范围均出现了大幅度的减小,如当4%ITO纳米粒子修饰g-C3N4颗粒后其发光范围窄至450~500 nm且荧光强度急剧减弱,究其原因,可能是由于ITO/g-C3N4界面处电子发生耦合作用,致使g-C3N4颗粒表面产生的光生电子转移到ITO颗粒表面,从而抑制了电子与空穴的复合[17]。

图7 x%ITO/g-C3N4(x=0,2,3,4)的室温荧光光谱图Fig.7 Room temperature fluorescence spectra for x%ITO/g-C3N4(x=0,2,3,4)
2.5 光催化性能表征
x%ITO/g-C3N4(x=0,1,2,3,4)催化剂光解水析氢性能受光照时间影响,其关系如图8所示。从图8中可知:(1)x%ITO/g-C3N4(x=0,1,2,3,4)催化剂光解水析氢性能与光照时间成近线性关系;(2)随着ITO附载量的增多,g-C3N4催化剂光解水析氢能力得到显著提高,纯g-C3N4催化剂光解水析氢速率为120~150 μmol·g-1·h-1[4];附载 1%ITO 后,g-C3N4催化剂光解水析氢速率提高到 250 μmol·g-1·h-1;进一步附载ITO后 (4%),g-C3N4催化剂光解水析氢速率增加变缓,但也达到 350 μmol·g-1·h-1,说明,附载ITO能有效提高g-C3N4光催化剂活性。

图8 x%ITO/g-C3N4(x=0,1,2,3,4)催化剂光解水析氢性能与光照时间关系图Fig.8 Dependence of H2 evolution performance on irradiation time over x%ITO/g-C3N4(x=0,1,2,3,4)
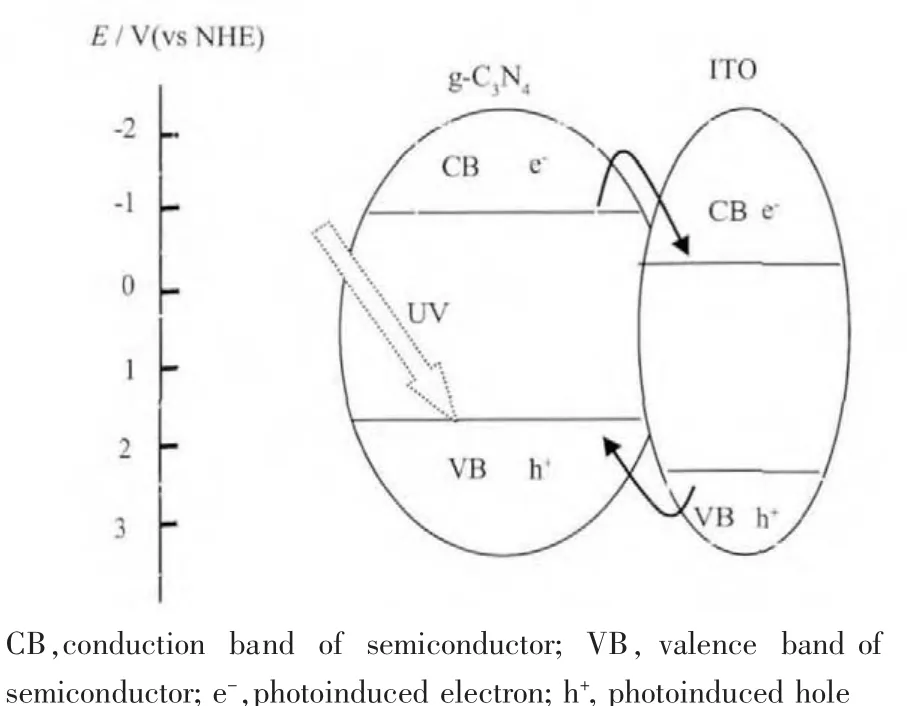
图9 UV-Vis光照射下ITO/g-C3N4异质结催化剂电子转移示意图Fig.9 Schematic diagram of the charge transfer and separation in the ITO/g-C3N4 photocatalyst under UV-Vis irradiation
一些理论计算和实验结果表明,g-C3N4具有非常合适的带边电位,其导带和价带电位分别为-1.3 eV 和+1.4 eV(vs NHE,pH=7),满足光解水产氢的热力学要求[18-19],但是大量的实验结果也表明,其光解水产氢速率较低,究其原因,是由于g-C3N4可见光吸收范围较窄,同时其电子迁移率较低。一个性能良好的光催化剂的首要条件是能较好地吸收可见光,并能有效地将其转化为光生电子,其次是这些光生电子能为光催化剂所捕获以抑制光生载流子间的复合。ITO具有良好的导电能力,带隙约为2.8 eV,但是其光解水产氢性能很弱[20]。当少量的ITO(In2O3导带电位约为-0.6 eV[21])附载于g-C3N4颗粒表面后,g-C3N4半导体光照后受激产生的电子在电势作用下会迁移到ITO表面上,实现了光生电子在g-C3N4与ITO两相间相互分离,有效提高了催化剂的量子效率,其电子转移示意图如图9所示。聚集在g-C3N4表面的光生空穴会为溶液中的乙醇牺牲试剂所捕获,抑制了光生截流子的复合,提高了g-C3N4光催化剂析氢活性。因此,适量附载ITO纳米粒子至g-C3N4颗粒表面,可有效提高g-C3N4的光催化析氢性能。
3 结 论
以三聚氰胺、硝酸铟和四氯化锡为原料,采用低温热分解法分别制备出g-C3N4粉体和ITO纳米粒子,通过静电引力作用将少量ITO纳米粒子负载于g-C3N4粉体颗粒表面,制成ITO/g-C3N4异质结光催化剂,借助XRD、SEM、TEM、UV-Vis等手段对样品进行了表征,讨论了ITO附载量对g-C3N4光催化析氢活性的影响。结果表明:
(1)少量ITO纳米粒子可与g-C3N4形成异质结光催化剂,在可见光模拟系统中ITO能显著提高g-C3N4光催化析氢活性;
(2)当ITO附载量为4%时,g-C3N4光解水析氢速率可达到 350 μmol·g-1·h-1左右。
[1]Shalom M,Inal S,Fettkenhauer C,et al.J.Am.Chem.Soc.,2013,135:7118-7121
[2]Zhao X D,Ma Z N,Wu D H,et al.Int.J.Hydrogen Energy,2015,40:8897-8902
[3]Zhang Y W,Liu J H,Wu G,et al.Nanoscale,2012,4:5300-5303
[4]Hong JD,Xia X Y,Wang Y S,et al.J.Mater.Chem.,2012,22:15006-15012
[5]Zhang Y,Mori T,Ye J.J.Am.Chem.Soc.,2010,132:6294-6295
[6]Yue B,Li Q,Iwai H,et al.Adv.Mater.,2011,12:034401-034405
[7]Wang X,Chen X,Thomas A,et al.Adv.Mater.,2009,21:1609-1612
[8]Ge L,Han C C.Appl.Catal.B:Environ.,2012,117~118:268-274
[9]ZHAO Xue Guo(赵学国),HUANG Zu Zhi(黄祖志).Chinese J.Inorg.Chem.(无机化学学报),2015,31:69-73
[10]Yan H,Yang H.J.Alloys Compd.,2011,509:L26-9
[11]Wang Y,Shi R,Lin J.Energy Environ.Sci.,2011,4:2922-2929
[12]Suryawanshi A,Dhanasekaran P,Mhamane D,et al.Int.J.Hydrogen Energy,2012,37:9584-9589
[13]Xiang Q,Yu J.J.Phys.Chem.C,2011,115:7355-7363
[14]Marı′n-Ramos P,Marı′n-Gil J,Dante R C,et al.Int.J.Hydrogen Energy,2015,40:7272-7281
[15]Jiang D L,Chen L L,Zhu JJ,et al.Dalton Trans.,2013,42:15726-15734
[16]Zhu H L,Yao K H,Zhang H,et al.J.Phy.Chem.B,2005,109:20676-20679
[17]Liu J,Liu Y,Liu N Y,et al.Science,2015,347:970-974
[18]Yang S,Gong Y,Zhang J,et al.Adv.Mater.,2013,25:2452-2456
[19]Ge L,Han C,Xiao X,et al.Int.J.Hydrogen Energy,2013,38(17):6960-6969
[20]Xu L L,Guan J G,Gao L,et al.Catal.Commun.,2011,12:548-552
[21]Chen S F,Yu X L,Zhang H Y,et al.J.Hazard.Mater.,2010,180:735-740
猜你喜欢
中国粉体技术(2022年2期)2022-03-19 08:29:28
——潘桂棠光生的地质情怀
沉积与特提斯地质(2021年2期)2021-07-20 06:33:26
粉末冶金技术(2021年1期)2021-03-29 02:35:10
粉末冶金技术(2021年1期)2021-03-29 02:34:48
陶瓷学报(2019年6期)2019-10-27 01:18:18
物理化学学报(2017年3期)2017-03-11 00:25:30
浙江农业科学(2016年11期)2016-05-04 04:16:45
昭通学院学报(2016年5期)2016-02-24 10:51:12
石家庄铁道大学学报(自然科学版)(2015年3期)2015-02-28 15:05:43
应用化工(2014年11期)2014-08-16 15:59:13