
乙炔氢氯化反应制备氯乙烯低固汞催化剂失活机理
2015-09-14 05:42:48支林轩唐浩东刘化章裴文俊
化学反应工程与工艺 2015年4期
支林轩,李 瑛,唐浩东,刘化章,裴文俊
乙炔氢氯化反应制备氯乙烯低固汞催化剂失活机理
支林轩1,李 瑛1,唐浩东1,刘化章1,裴文俊2
1.浙江工业大学工业催化研究所,浙江 杭州 310032;2.云南契合投资有限公司,云南 昆明 650000
采用氮气吸附脱附技术、扫描电镜(SEM)、程序升温脱附-质谱联用(TPD-MS)、程序升温氧化-质谱联用(TPO-MS)和热重分析仪(TG)等对新鲜及反应800h后的乙炔氢氯化低固汞催化剂进行了表征及失活机理分析。结果表明,反应后的低固汞催化剂的HgCl2的流失仅为1.6%,积炭量为13.5%,并测得含硫化合物,说明硫中毒和积炭是催化剂失活的主要原因。
乙炔氢氯化 氯乙烯 低固汞催化剂 失活机理 积炭 中毒
聚氯乙烯(PVC)为五大通用树脂之一,具有广阔的市场需求[1]。聚氯乙烯的单体氯乙烯(VCM)可由多条工艺路线合成,常用的有电石法和乙烯法两种[2,3],在我国,由于国内资源特点所限,PVC生产线以电石法为主[4]。目前电石法所用的高汞催化剂虽然性能优异,成本低廉,但是在工业生产过程中HgCl2流失严重,易造成严重的环境污染及资源浪费,涉汞催化剂的环保问题一直是该体系发展的瓶颈,是现阶段电石法生产聚氯乙烯行业发展亟待解决的问题。而无汞催化剂的研发虽然历经多年的研究,一直存在着性能和成本无法兼顾的困局[5],目前尚处于实验室阶段。为此,国家工信部于2010年6月4日发布了“电石法聚氯乙烯行业汞污染综合防治方案”,要求全行业在2015年全面采用低固汞催化剂。按照国标要求,低固汞催化剂的活性组分氯化汞的负载量在4%~6.5%,比高汞催化剂(10%~12%)降低了近一半,因此对催化剂的性能提出了更高的要求。近几年来,虽然已经有一些低固汞催化剂的试用报道[6-8],但大多来自企业的性能评估,对低固汞催化剂的失活机理研究鲜有报道,有关hgCl2催化剂失活方面的研究报道较少,且集中在上世纪 80年代之前[[9-12],主要是针对高汞催化剂的失活机理,低固汞催化剂除了氯化汞负载量降低一半之外,而且多采用多元配方[13],失活机理有可能发生改变,因此开展低固汞催化剂失活机理的研究对解决汞流失引起的环保问题至关重要。
由于乙炔氢氯化反应的特殊性,低固汞催化剂的失活原因主要包括以下几点:(1)活性组分的挥发:HgCl2为共价型化合物,沸点276 ℃,且蒸汽压随温度升高而急骤升高,常温时微量挥发,140 ℃以上则变得十分明显。乙炔的氢氯化反应为强放热反应(25 ℃条件下,rmHθΔ为 -124.8 kJ/mol),在固定床反应中,反应热点温度往往超过180 ℃,极易造成HgCl2挥发。(2)该反应中活性组分为HgCl2,而非单质汞,若反应物乙炔过量,还可能发生HgCl2与乙炔还原成单质汞而引起的失活。(3)乙炔气中的有害杂质(H2S和H3P)能与催化剂发生不可逆吸附,使催化剂中毒,同时杂质还能与HgCl2反应,生成无活性的汞盐[12];(4)原料乙炔和产物氯乙烯在活性炭表面可能发生分子间聚合,沉积物沉积在载体表面堵塞孔道,阻碍了反应进行,造成反应活性下降[13]。(5)催化剂破碎等。
本工作以活性炭为载体,采用浸渍法制备了复方低固汞催化剂,在模拟工业生产的条件下稳定运行超过800h,考察该催化剂乙炔氢氯化反应活性。同时,将反应前后的催化剂进行了表征,从活性组分流失、积炭和中毒3个方面对复方低固汞催化剂的失活原因进行分析,探究提高催化剂活性及稳定性的方法。对新型低固汞及无汞催化剂的开发、工业应用及进一步减少氯乙烯行业的汞污染都具有重要的意义。
1 实验部分
1.1低固汞催化剂制备及HgCl2负载量分析
采用浸渍法制备催化剂。称取一定量的HgCl2和氯化物助剂溶解于100 mL浓度为2 mol/L的盐酸溶液中,待溶解后将此浸渍液平均分成3份,称取10 g粒径为1~2 mm的活性炭(AC),分3次加入浸渍液并干燥,每次室温浸渍18h,120 ℃干燥10h。重复3次后制得新鲜催化剂,记为HC。
HgCl2负载量分析采用消解-铜试剂滴定法,具体实验方法参见《中华人民共和国化工行业标准(HG/T 4192-2011):氯乙烯合成用低固汞触媒》。
1.2 催化剂活性评价
活性评价实验在固定床连续流动反应器中进行,反应管为内径12 mm的玻璃管。将5.0 mL催化剂装入反应器中,在氮气保护下(20 mL/min)以10 ℃/min升温至140 ℃,保持0.5h后,切换成氯化氢气体,于140 ℃活化1h后,切换成乙炔和氯化氢的混合气体进行反应。乙炔和氯化氢体积比为1∶1.1,乙炔气体流量为3 mL/min(0.15 MPa),氯化氢气体流量为3.3 mL/min(0.3 MPa)。乙炔体积空速(GHSV)为36h-1。反应后气体的过量氯化氢经氢氧化钠吸收后用六通阀进样,用气相色谱采用面积归一化法分析尾气组成,乙炔的转化率(XA)及氯乙烯的选择性(SVCM)分别按照以下公式计算:

式中:φA为剩余乙炔的体积分数;φVCM为氯乙烯的体积分数。
催速实验反应条件:催化剂装填量为2.0 mL,温度180 ℃,压力0.15 MPa,空速1 000h-1。
1.3 催化剂表征
材料比表面积和孔径分布的测定在美国康塔公司的Autosorb-IQ型吸附仪上进行,样品先在300 ℃抽空至1.33×10-4Pa,保持3h以脱除样品中物理吸附的水分,然后在-196 ℃根据静态法测量吸-脱附等温线。由BET法计算材料的比表面积,用DFT法计算孔分布。
样品的程序升温脱附/程序升温氧化-质谱联用(TPD/TPO-MS)实验在自行组装的装置中进行。称取约0.03 g样品置于U型石英管中,样品分析前,先在110 ℃下恒温2h以脱去物理水,待基线平稳后,以10 ℃/min的升温速率从110 ℃升至 800 ℃。TPD-MS中Ar流量30 mL/min,TPO-MS中空气流量30 mL/min。尾气用Hiden Analytical Ltd生产的QIC-20质谱进行在线检测。检测信号为质荷比(m/e)为28,44和64。
样品的热重(TG-DTG)表征在Netzsch-STA449C热分析仪上进行,样品装填量10 mg,粒度为1.0~1.4 mm(10~18目)。样品在110 ℃恒温2h以脱去物理水,气体流速30 mL/min,再以升温速率10 ℃/min升温至800 ℃。
采用Hitachi S-4700Ⅱ型扫描电子显微镜(SEM)表征样品的形貌,工作电压15 kV,放大倍数为3 000倍,元素含量采用扫描仪自带的能谱仪(EDS)进行分析。
2 结果与讨论
2.1 催化剂性能评价
图 1为催化剂的乙炔转化率及氯乙烯选择性随时间的变化趋势。由图可知,实验室自制的低固汞催化剂的初活性较好,乙炔转化率在97%以上,氯乙烯的选择性大于99%,转化率随着反应的进行而缓慢下降。当反应时间超过800h,转化率降至90%,而氯乙烯的选择性维持在99%以上。实验结束后,在氮气保护下降至室温后取出样品,称取反应后催化剂的质量和新鲜催化剂一起作表征测试,研究催化剂失活原因。
2.2低固汞催化剂的表征
2.2.1 反应前后催化剂质量及HgCl2负载量变化
反应前后催化剂的质量及HgCl2的负载量变化见表1。由表可知,反应前催化剂的质量为2.08 g,反应800h后,催化剂的质量为2.36 g,反应后催化剂的增重率为13.5%。为了分析催化剂增重原因,采用EDS对新鲜的(HC)和反应后的(HC-used)催化剂进行了表面元素含量测定,结果如表1所示。另外,由于高真空测试条件下,HgCl2有挥发,因此HgCl2的含量无法准确给出。由表可知,反应后Cl元素的含量从5.9%增加到11.1%,说明含氯的产物氯乙烯或者其他副产物富集到了催化剂表面,造成了催化剂的增重。增重也有可能是积炭引起的,但是由于 EDS对炭的含量分析不准确,无法对积炭进行定量分析。除氯元素含量增加之外,催化剂增加了0.9%的S元素,可能是由乙炔气体的微量含硫物质随着反应的进行富集到催化剂表面。
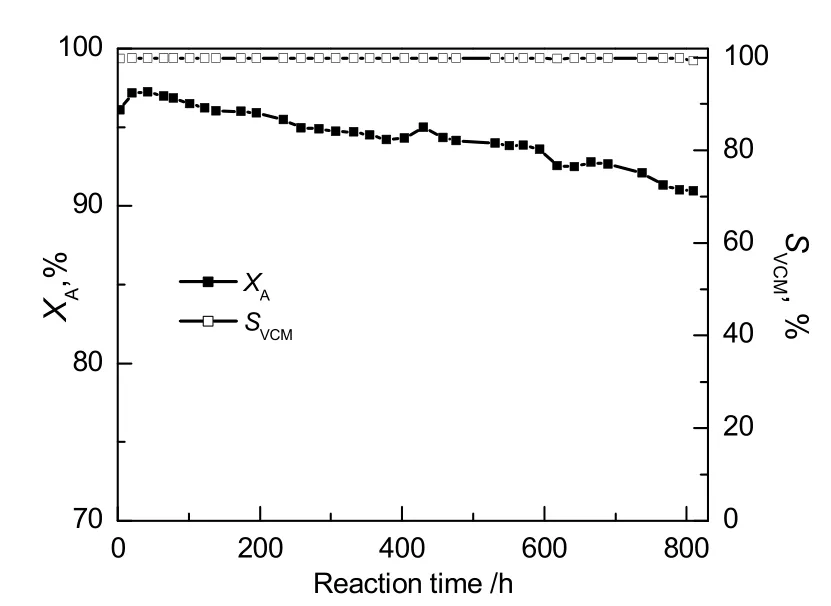
图1 低固汞催化剂的乙炔氢氯化反应性能Fig.1 Catalytic performance ofhC catalyst in acetylenehydrochlorination
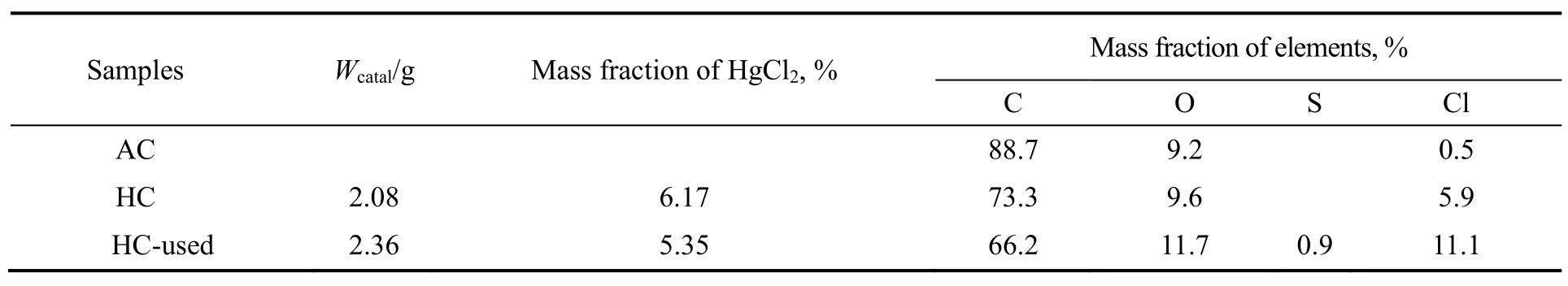
表1 活性炭载体及反应前后的低固汞催化剂物性及元素含量变化Table 1 Physical properties and elemental compositions of activated carbon, fresh and deactivated catalyst
用铜试剂滴定法[16,17]测定反应前后催化剂中HgCl2含量分别为6.17%和5.35%,根据新鲜催化剂中的HgCl2负载量和反应800h后催化剂的氯化汞的负载量计算HgCl2的损失率仅为1.6%,说明该条件下催化剂HgCl2的流失并不是很严重。反应后HgCl2负载量的降低主要是由于反应后的催化剂的重量增加造成的,并不是HgCl2的挥发引起的。按照工业报道的数据,高汞催化剂使用后HgCl2流失大于60%[6,8],这也是工业应用情况下高汞催化剂中的hgCl2的流失是造成催化剂失活的主要原因[13,18],但是对低固汞催化剂而言,由于其HgCl2负载量远远低于活性炭对HgCl2的饱和吸附量,在工业反应器串联使用的情况下,一段的HgCl2挥发进入气相后还会被二段反应器中的低固汞催化剂吸附截留[18],因此,HgCl2的挥发造成的催化剂活性下降影响远不如高汞催化剂大。
2.2.2 反应前后催化剂的孔结构
表2给出反应前后催化剂及活性炭载体的孔结构参数。由表可知,引入活性组分及助剂后,催化剂的比表面积和孔容均有所下降。
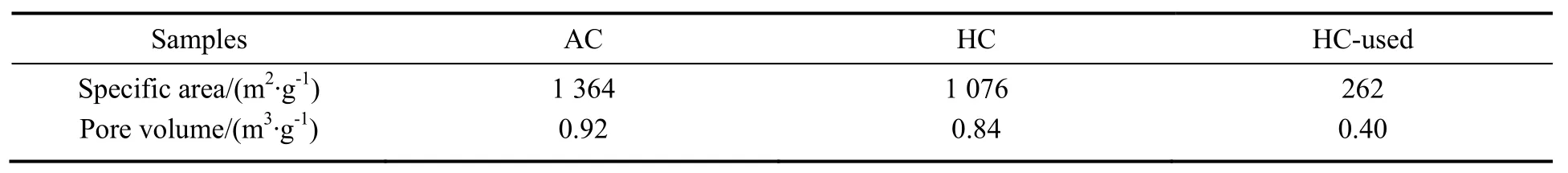
表2 活性炭载体及反应前后的低固汞催化剂物性Table 2 Physical properties of activated carbon, fresh and deactivated catalyst
图2为反应前后催化剂及载体活性炭的吸脱附等温线及孔径分布图。由图可知:载体AC的吸附-脱附等温线的形状介于Ⅰ型和Ⅳ型等温吸附线之间,在较低的相对压力(P/P0小于0.1)下吸附量迅速上升,这反映的是微孔填充现象;在中等的相对压力下,由于毛细凝聚的发生,出现了h4型回滞环[20,24],这说明存在中孔和微孔混合孔结构。新鲜催化剂和活性炭载体相比,吸附等温线的形状没有发生较大的改变,只是吸附量降低,但是从DFT孔径分布图可看出,微孔孔径从0.56nm增加到0.78nm,而大于1nm的孔分布基本没有变化。反应后催化剂的吸脱附等温线和反应前的吸附等温线存在明显差异。不但H4型的滞后环消失,而且低压下对应于微孔的部分拐点几乎消失,类似于Ⅱ型等温线。从图2还可以看出,反应前后的催化剂孔径分布位置差别不大,均存在大小不一的微孔和中孔,主要集中在3.5,1.4和0.6nm,但峰强度降低较明显。说明反应后的催化剂(HC-used)孔结构被乙炔及产物氯乙烯自聚或反应过程中产生的积炭物种堵塞,微孔和中孔大量减少,催化剂的活性位被积炭物种所覆盖,影响了原料气与活性中心的接触,从而导致反应活性下降。
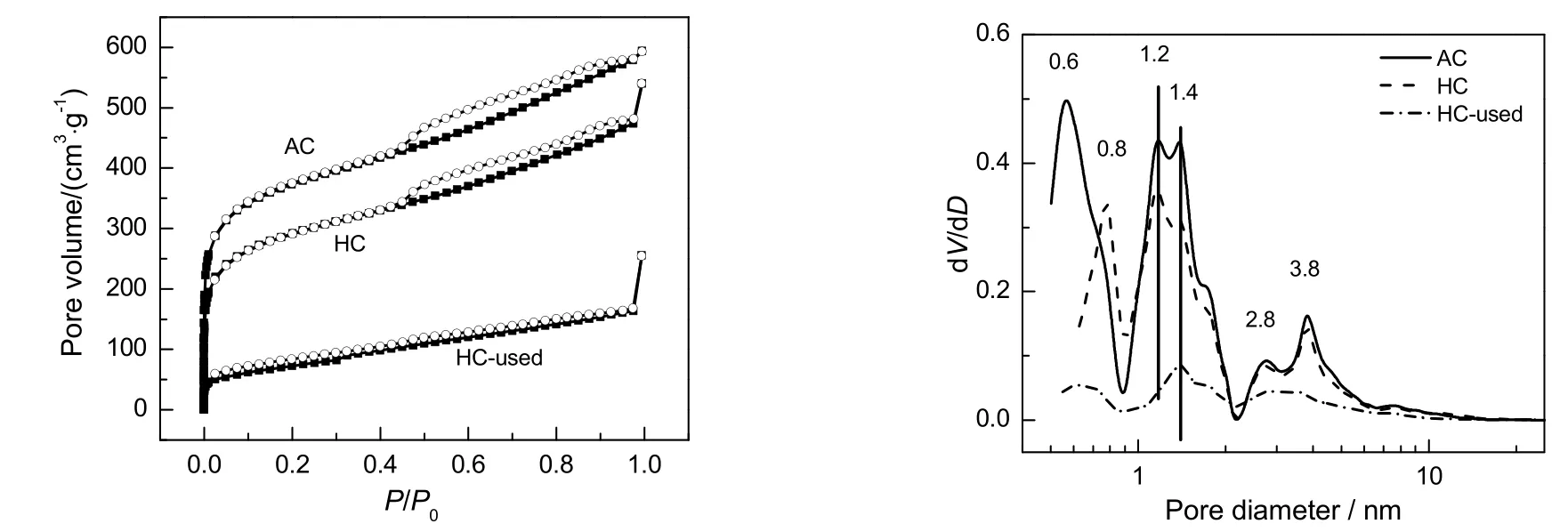
图2 活性炭载体、新鲜催化剂及反应后催化剂的吸附-脱附等温线及孔径分布Fig.2 N2adsorption-desorption isotherms and pore size distributions of AC,hC andhC-used
图3为新鲜和反应后的催化剂的扫描电镜图。由图3可知,新鲜的HC催化剂存在明显蜂窝状孔道,尺寸在1~10 μm,除存在少数细碎颗粒外,孔道较为干净。由于扫描电镜的放大倍数不够,看不到纳米的孔道。反应后催化剂形貌发生了明显变化,微孔孔道中存在较多絮状及颗粒状杂质,几乎被填满,有可能是积炭造成的[18,22-24]。反应过程中乙炔及产物氯乙烯自聚或反应过程中产生的积炭物种,使得微孔和中孔大量减少,催化剂的活性位被积炭物种所覆盖,影响了反应物与活性中心的接触,从而导致反应活性下降。上述结果验证了氮气吸附测定的结果。

图3 新鲜和反应后催化剂的SEM照片Fig.3 SEM images ofhC andhC-used catalysts
2.2.3 反应前后催化剂的表面性质研究
在惰性气氛下对样品进行程序升温脱附,用热重检测样品的质量变化,可以用于分析催化剂表面含碳物种沉积程度[25-27],用气相质谱检测尾气中脱附出来的气体信号,可以关联样品的表面化学性质及积炭情况。图4为活性炭载体、新鲜催化剂和反应后的催化剂的Ar-TG/DTG图。由图可知,样品的失重主要分为3个阶段,其中在110~200 ℃失重主要是由于样品中少量水分挥发引起。
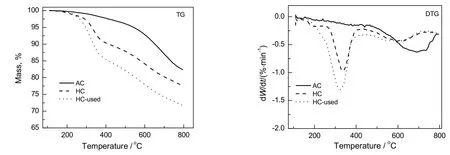
图4 AC,HC和HC-used样品在氩气气氛下的热重图谱Fig 4 TG and DTG curves of AC,HC andhC-used catalyst in argon flow
表3为在Ar气氛下的TG/DTG实验数据。由表可知,与活性炭载体相比,新鲜催化剂在350 ℃左右的失重量约为9.0%,扣除催化剂的少量水失重(2.1%),失重量约为6.9%,这一数值与HgCl2的负载量比较接近,说明该区间的失重主要是由HgCl2的挥发引起的[28,29]。
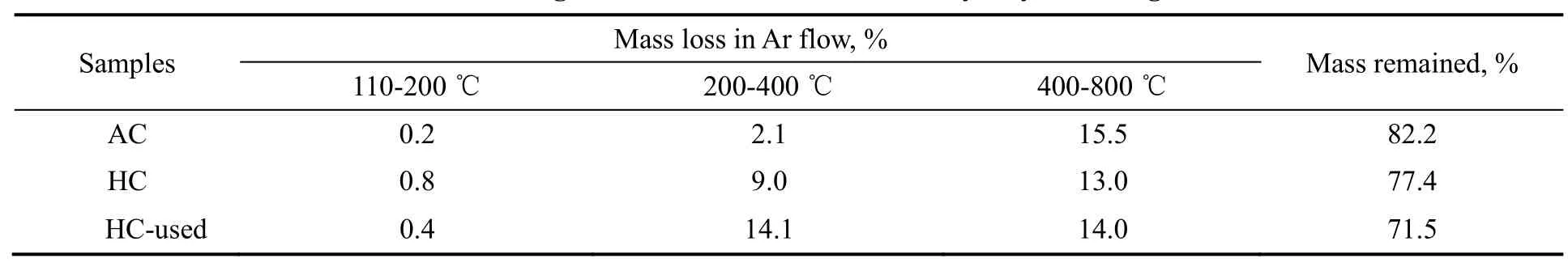
表3 AC,HC和HC-used催化剂在氩气气氛下的质量变化Table 3 Mass changes of AC,hC andhC-used catalyst by TG in argon flow
图5为Ar气氛下活性炭载体、新鲜催化剂及反应后催化剂的TPD-MS图。由图可知,新鲜催化剂与活性炭载体在250~450 ℃的CO和CO2脱附曲线相似,新鲜催化剂上第1个CO2脱附峰的温度(325 ℃)比载体的(400 ℃)低了75 ℃。因此推断,这部分失重主要是HgCl2的挥发以及少量样品中活性炭载体的表面基团分解失重引起的。反应后的催化剂在 350 ℃前后的失重量比新鲜催化剂增加了5.1%,这部分失重是由于部分反应后沉积的物种的脱附引起的。根据图5可知,反应后的催化剂分别在235和310 ℃位置出现CO脱附峰,在325 ℃的CO2脱附峰强度高于新鲜催化剂,说明反应后的催化剂上表面沉积了较多的炭物种。在200~800 ℃的失重是由活性炭载体的表面基团分解引起。活性炭的 CO脱附峰出现在 670 ℃,对应于活性炭表面的酚羟基、羰基和醌基的分解,CO2的脱附峰分别出现在400和575 ℃,400 ℃左右出现的CO2脱附峰归属为活性炭表面的羧基的分解,而在 575 ℃左右出现的 CO2脱附峰归属为活性炭表面的酸酐和内酯基的分解[30-32]。新鲜催化剂的Ar-TPD-MS脱附峰CO和CO2信号和活性炭载体相比,均出现了较大的变化,首先是670 ℃的CO脱附峰转化成了635和820 ℃两个峰,说明活性炭的表面基团和负载组分之间具有一定的相互作用,具体解释还需要进一步的研究证明。CO2的脱附峰也发生了一定的变化,新鲜催化剂的CO2脱附峰出现了3个峰,分别在325,575和780 ℃,其中325 ℃的脱附可能对应于活性炭表面羧基的分解脱附峰(400 ℃)。由于活性组分和活性炭载体之间的相互作用,分解温度提前,而575 ℃处对应于活性炭表面的酸酐和内酯基的分解峰没有改变。有些文献把 780 ℃的脱附峰归属为内酯基的分解[33],这一点还需要进一步的研究证实。
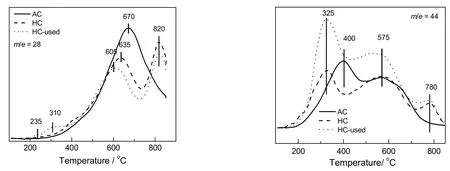
图5 Ar气氛下活性炭载体、新鲜催化剂及反应后催化剂的TPD-MS图谱Fig.5 TPD-MS profiles of AC,hC andhC-used catalysts in argon flow
为了进一步研究反应后的催化剂上的积炭物种,在空气气氛下对载体、新鲜催化剂及反应后催化剂做了热重实验,在空气下进行程序升温氧化-质谱联用(TPO-MS)实验,结果见图6,图7和表4。
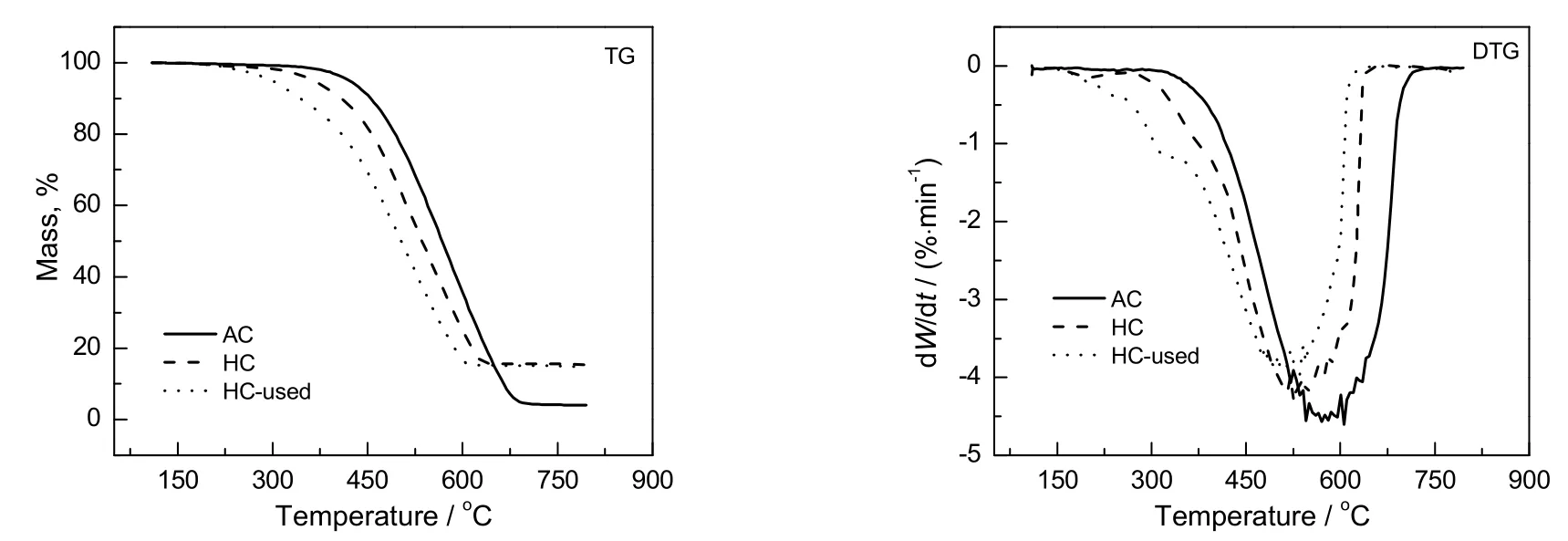
图6 样品在空气气氛下的热重分析曲线Fig.6 TG and DTG curves of AC,HC andhC-used catalysts in air flow

表4 AC,HC和HC-used在空气气氛下的质量变化Table 4 Mass change of AC,hC andhC-used catalyst in air flow
从表4和图6可知,3个样品的热重分析曲线主要区别在200~400 ℃,其中活性炭和新鲜催化剂失重量与氩气气氛中的结果基本相同,反应后催化剂的失重量由14.1%增加到17.6%。这是由于表面难以挥发的积炭物种被氧气氧化燃烧掉了,这部分难以挥发的积炭物种的失重量约为3.5%。加上Ar气氛下相同温度区间易挥发的积炭物种的失重量 5.1%,则反应后沉积在催化剂表面的沉积物种在110~400 ℃之前失重量约为8.6%,略小于反应后催化剂增重量(13.5%),说明还有部分积炭氧化温度高于400 ℃。

图7 空气气氛下活性炭载体、新鲜催化剂及反应后催化剂的TPO-MS图谱Fig.7 TPO-MS profiles of AC,HC andhC-used catalysts in air flow
由图7可知,在200~350 ℃,反应后催化剂的CO信号峰强度明显高于新鲜催化剂,说明反应催化剂上存在积炭物种,在400~700 ℃主要出现CO和CO2的信号峰,其中CO的信号峰更强,这是由于空气气氛下,炭的不完全燃烧造成的。由图7(a)可知,活性炭载体、新鲜催化剂及反应后的催化剂燃烧产生的CO信号主峰分别出现在540,600和710 ℃,反应后的催化剂的CO起峰温度介于新鲜催化剂和载体之间。由于空气气氛下的程序升温氧化实验得到的气体以CO为主,说明该实验中样品没有充分燃烧,较难区分表面的积炭物种及载体活性炭的燃烧温度,另外还做了纯氧气氛下的程序升温氧化实验,但起峰温度在400~600 ℃,也很难区分表面积炭和载体活性炭的燃烧温度,因此程序升温氧化的产物检测结果只能说明表面存在积炭物种,并影响了载体的燃烧,但较难进行定量。图7(c)给出SO2(m/e为64)的信号峰,可以看出,失活的催化剂在240和340 ℃出现了明显的SO2的峰,而新鲜催化剂和AC载体则没有SO2的峰,说明失活催化剂上确实存在含硫的物种。
综合惰性气氛及空气气氛下热重分析、程序升温脱附及在线质谱检测的结果,可发现催化剂的积炭物质的分解和HgCl2的挥发造成的失重主要发生在200~400 ℃,反应后的催化剂存在明显积炭现象,积炭物质分解温度起始于200 ℃,惰性气氛下分解失重为5%左右,空气气氛下燃烧失重大约为9%~10%。
2.3h2S中毒实验
结合EDS和TPO-MS结果表明反应后的催化剂表面确实存在含硫化合物,这些含硫化合物的来源虽然受仪器和实验方法限制未能检测出来,但很可能是由乙炔原料气中痕量含硫物质或原料气处理过程引入催化剂表面累积造成,或制备催化剂的原料中微量硫化合物带来的。
为验证H2S对催化剂性能的影响,将新鲜催化剂进行了预硫化处理:将一定量新鲜催化剂在H2S气氛下140 ℃处理3h,记为HC-S。将HC和HC-S在180 ℃和空速为1 000h-1的条件下进行反应,结果如图8。由图可知,H2S的预毒化使新鲜催化剂的活性显著下降,乙炔转化率从33%降低至25%。说明H2S处理后的低固汞催化剂性能下降明显,因此在评价过程中,使用的原料乙炔气即使含有微量H2S,亦会直接影响催化剂活性。因此在氯乙烯的工业生产中,对乙炔中的H2S含量有非常严格的控制,在乙炔生产中需要引入净化工艺[1,15,22]。
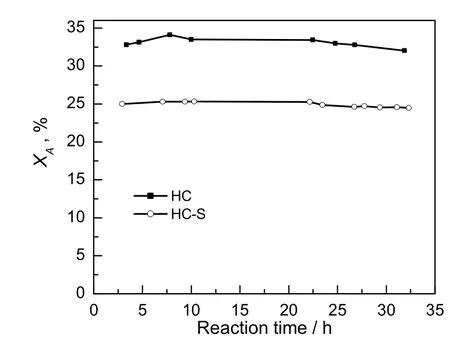
图8 HC和HC-S催化剂的乙炔氢氯化反应性能Fig.8 Catalytic performance ofhC andhC-S catalyst for acetylenehydrochlorination
3 结 论
以活性炭为载体,采用浸渍法制备复方低固汞催化剂,在模拟工业生产的条件下稳定运行超过800h,考察了该催化剂的乙炔氢氯化反应性能及失活机理,发现低固汞催化剂的失活速率较慢,其失活的主要原因是由于催化剂的积炭及硫中毒,而氯化汞的挥发并不是很严重,在体积空速36h-1及140 ℃的条件下,反应800h后,低固汞催化剂的HgCl2损失率仅为1.6%,即HgCl2的挥发损失已经不是低固汞催化剂失活的主导原因,反应过程的积炭以及硫中毒是该催化剂的失活的主要原因。
[1]邴涓林, 黄志明. 聚氯乙烯工艺技术[M]. 北京: 化学工业出版社, 2007: 2-13.
[2]张 晶, 段明哲, 张志刚, 等. 氯乙烯生产技术[J]. 化工进展, 2014, 33(12): 3164-3169. Zhang Jing, Duan Mingzhe, Zhang Zhigang, et al. Vinyl chloride monomer production technology[J]. Chemical Industry and Engineering Progress, 2014, 33(12): 3164-3169.
[3]Wilkes C E, Summers J W, Daniels C A. 聚氯乙烯手册[M]. 乔辉, 译. 北京: 化学工业出版社, 2008: 22.
[4]张英民, 郎需霞, 梁锡伟. 电石法PVC行业发展趋势及构想[J]. 中国氯碱, 2012, (3): 1-5. Zhang Yingmin, Lang Xuxia, Liang Xiwei. Developing trend and outlook of PVC industry of calcium carbide method[J]. China Chlor-Alkali, 2012, (3): 1-5.
[5]张学鲁, 周 军, 李国栋. 低固汞催化剂应用体系研发进展[J]. 中国氯碱, 2013, (7): 17-19. Zhang Xuelu, Zhou Jun, Li Guodong. Progress on the research of low-immobilized mercury catalyst application system[J]. China Chlor-Alkali, 2013, (7): 17-19.
[6]杨超松, 周 菊. 低汞触媒应用总结[J]. 化工技术与开发, 2012,41(8): 59-60. Yang Chaosong, Zhou Ju. Application summary of low-mercury catalyst[J]. Technology & Development of Chemical Industry, 2012,41(8): 59-60.
[7]熊 磊, 张 明. 低汞触媒试用总结[J]. 中国氯碱, 2014, (11): 22-24. Xiong Lei, Zhang Ming. Summary of low mercury catalyst trial [J]. China Chlor-Alkali, 2014, (11): 22-24.
[8]周 军, 张学鲁, 李春华. 聚氯乙烯低汞化实践与总结[J]. 中国氯碱, 2014, (7): 15-18. Zhou Jun, Zhang Xuelu, Li Chunhua. Polyvinyl chloride low mercury practice and summary[J]. China Chlor-Alkali, 2014, (7):15-18.
[9]赵振兴, 刘大壮. 升华型催化剂活性组分的升华机理与流失动力学[J]. 郑州大学学报(工学版), 1991, 12(2): 1-9. Zhao Zhenxing, Liu Dazhuang. The loss mechanism and kinetics by sublimation of active species on subliming catalyst[J]. Journal of Zhengzhou Institute of Technology, 1991, 12(2): 1-9.
[10]田 祎, 邢 奕, 刘景洋, 等. 氯化汞触媒的汞挥发速率[J]. 北京科技大学学报, 2012, 34(8): 939-942. Tian Yi, Xing Yi, Liu Jingyang, et al. Mercury evaporation rate of mercury chloride catalysts[J]. Journal of University of Science and Technology Beijing, 2012, 34(8): 939-942.
[11]Agnew J B, Shankarh S. Catalyst deactivation in acetylenehydrochlorination[J]. Industrial and Engineering Chemistry Product Research and Development, 1986, 25(1): 19-22.
[12]Shankarh S, Agnew J B. Kinetics of acetylenehydrochlorination[J]. Industrial and Engineering Chemistry Product Research and Development, 1980, 19(1/2): 232-237.
[13]王鹏程,杨雪超,李瑛,多元配方乙炔氢氯化反应催化剂研究进展[J]. 聚氯乙烯,2015,43(4): 1-8. Wang Pengcheng, Yang Xuechao, Li Ying. Research progress in multiple catalysts for acetylenehydrochlorination [J]. Polyvinyl Chloride, 2015, 43(4): 1-8.
[14]郑石子, 颜才南, 胡志宏, 等. 聚氯乙烯生产与操作[M]. 北京: 化学工业出版社, 2007: 208.
[15]徐龙龙, 王绪根, 张海洋, 等. 一种新型炭载体在乙炔氢氯化反应中的应用[J]. 化工进展, 2011, 30(3): 536-541. Xu Longlong, Wang Xugen, Zhanghaiyang, et al. Application of a novel carbon carrier in acetylenehydrochlorination[J]. Chemical Industry and Engineering Progress, 2011, 30(3): 536-541.
[16]蒋万安, 李 浩. 用铜试剂法测定氯化汞含量[J]. 氯碱工业, 1993, 9: 42-44. Jiang Wanan, Lihao. Mercuric chloride content was measured by copper reagent method[J]. Chlor-Alkali Industry, 1993, 9: 42-44.
[17]龙 睿. 低汞触媒中氯化汞含量的测定[J]. 聚氯乙烯, 2012, 40(2): 32-33. Long Rui. Determination of mercuric chloride content in low-mercury catalysts[J]. Polyvinyl Chloride, 2012, 40(2): 32-33.
[18]陈志强, 马现英. 环保型低汞触媒的应用[J]. 中国氯碱, 2009, (6): 9-11. Chen Zhiqiang, Ma Xianying. Application of environmental low-mercury catalyst[J]. China Chlor-Alkali, 2009, (6): 9-11.
[19]邓云祥, 邹永匡, 孔宪祥, 等. 聚氯乙烯生产原理[M]. 北京: 科学出版社, 1982: 133-134.
[20]辛 勤, 罗孟飞. 现代催化研究方法[M]. 北京: 科学出版社, 2009: 15-16.
[21]Sing K S W, Everett Dh,haul R A W, et al. Reporting physisorption data for gas/solid sustems with secial rference to the dtermination of surface area and prosity[J]. Pure and Applied Chemistry, 1985, 57(4): 603-619.
[22]Zhangh Y, Daih, Li W, et al. Non-mercury catalytic acetylenehydrochlorination over spherical activated-carbon-supported Au-Co(III)-Cu(II) catalysts[J]. Journal of Catalysis, 2014, 316(1): 141-148.
[23]王声洁, 沈本贤, 赵基钢, 等. 乙炔氢氯化PdCl2/C催化剂失活原因分析[J]. 石油化工, 2009, 38(3): 249-253. Wang Shengjie, Shen Benxian, Zhao Jigang, et al. Deactivation of PdCl2/C catalyst inhydrochiorination of acetylene[J]. Petro Chemical Technology, 2009, 38(3): 249-253.
[24]Wang L, Wang F, Wang J D, et al.hydrochlorination of acetylene to vinyl chloride over Pd supported on zeolite Y[J]. Reaction Kinetics Mechanisms and Catalysis, 2013, 110: 187-194.
[25]Zhangh Y, Dai B, Wang X G, et al.hydrochlorination of acetylene to vinyl chloride monomer over bimetallic Au-La/SAC catalysts[J]. Journal of Industrial and Engineering Chemistry, 2012, 18(1): 49-54.
[26]Zhangh Y, Dai B, Wang X G, et al. Non-mercury catalytic acetylenehydrochlorination over bimetallic Au-Co(III)/SAC catalysts for vinyl chloride monomer production[J]. Green Chemistry, 2013, 15: 829-836.
[27]Zhang J L, Sheng W, Guo C L, et al. Acetylenehydrochlorination over bimetallic Ru-based catalysts[J]. RSC Advances, 2013, 3: 21062-21068.
[28]郑石子. 聚氯乙烯生产问答[M]. 北京: 化学工业出版社, 1986: 92.
[29]Hutchings G J, Grady D T. Effect of drying conditions on carbon supported mercuric chloride catalysts[J]. Applied Catalysis, 1985, 16: 411-415.
[30]Figueiredo J L, Pereira M F R, Freitas M M A, et al. Modification of the surface chemistry of activated carbons[J]. Carbon, 1999, 37(9): 1379-1389.
[31]Figueiredo J L, Pereira M F R, Freitas M M A, et al. Characterization of active sites on carbon catalysts[J]. Industrial and Engineering Chemistry Research, 2007, 46: 4110-4115.
[32]冯国全, 蓝国钧, 李 瑛, 等. 硝酸水热处理活性炭对其负载的Ba-Ru-K氨合成催化剂性能的影响[J]. 催化学报, 2012, 33(7): 1191-1197. Feng Guoquan, Lan Guojun, Li Ying, et al. Effect ofhydrothermal treatment of activated carbon by nitric acid on activity of Ba-Ru-K/AC catalyst for ammonia synthesis[J]. Chinese Journal of Catalysis, 2012, 33(7): 1191-1197.
[33]Morales-Torres S, Maldonado-Hódar F J, Pérez-Cadenas A F, et al. Design of low-temperature Pt-carbon combustion catalysts for VOC’s treatments[J]. Journal ofhazardous Materials, 2010, 183(1/3): 814-822.
Deactivation Mechanism of Low-Mercury Catalyst for Acetylenehydrochlorination
Zhi Linxuan1, Li Ying1, Tanghaodong1, Liuhuazhang1, Pei Wenjun2
1. Institute of Industrial Catalysis, College of Chemical Engineering, Zhejiang University of Technology,hangzhou 310032, China;2. Yunnan Qiehe Investment Co Ltd, Kunming 650000, China
The fresh and deactivated low-mercury catalysts were characterized by low-temperature nitrogen adsorption-desorption technique, scanning electron microscopy(SEM), temperature programmed desorptionmass spectrometry(TPD-MS), temperature programmed oxidation-mass spectrometry(TPO-MS) and thermal gravimetric analysis(TG). The results showed thathgCl2of deactivated catalyst lost by 1.6%, deposited carbon reached 13.5% and sulfocompounds were detected. The coking and sulfur poisoning were the main reasons for the new low-mercury catalyst.
acetylenehydrochlorination; vinyl chloride; low-mercury catalyst; deactivation; coking; poisoning
O643.38
A
1001—7631 ( 2015 ) 04—0343—09
2015-03-04;
2015-05-24。
支林轩(1990—),男,硕士研究生;李 瑛(1974—),女,教授,通讯联系人。E-mail: liying@zjut.edu.cn。
猜你喜欢
中国盐业(2018年16期)2018-12-23 02:08:28
汽车维护与修理(2018年7期)2018-10-13 06:03:56
劳动保护(2018年8期)2018-09-12 01:16:20
石油学报(石油加工)(2015年6期)2015-07-02 01:39:44
化学反应工程与工艺(2015年1期)2015-04-16 03:06:12
河南科技(2015年2期)2015-02-27 14:20:35
燃气轮机技术(2014年4期)2014-04-16 03:54:03
化工生产与技术(2014年4期)2014-02-27 13:41:51
食品科学(2013年19期)2013-03-11 18:27:17
微创医学(2012年6期)2012-01-13 03:47:38
