
渣油的分子组成模拟研究
2015-09-03 10:56任小甜申海平
石油炼制与化工 2015年11期
任小甜,阎 龙,申海平
(中国石化石油化工科学研究院,北京 100083)
渣油的分子组成模拟研究
任小甜,阎 龙,申海平
(中国石化石油化工科学研究院,北京 100083)
针对渣油中分子的结构组成,提出了一种基于结构导向集总的构建渣油分子的方法,选定65种单核分子和12种多核分子作为种子分子,以不同碳数的侧链进行划分,构建了共计3 749个虚拟分子来代表渣油。采用15种基本的结构向量组成的分子矩阵来描述每个分子的组成,可用于表示分子反应过程以及各分子物性的计算。以渣油中各分子特征分布为基础确定各分子摩尔分数的计算式,构建各物性计算值相对误差的目标函数,利用模拟退火算法计算渣油中各分子的相对含量。选取3种渣油进行分子建模研究,模拟计算结果表明,渣油各物性的计算值和实验测定值基本吻合,每种渣油芳香分的相对分子质量也呈现明显的γ分布。这3 749种分子可以基本反映渣油的整体性质,用分子特征分布的方法来计算渣油分子的相对含量是可行的。
渣油 结构向量 分子矩阵 特征分布 模拟退火算法
在炼油工业中,反应动力学模型以及过程模拟一直发挥着重要的作用。近年来,原油重质化的趋势不断加大,环保法规对油品的质量控制也不断提高,现有的集总反应动力学模型已经不能满足这样的要求,亟需要发展分子水平的详细动力学模型,从分子水平上认识石油组成,探究反应过程中的反应机理和分子转化规律。确定原料油的分子组成是建立分子尺度动力学模型的首要步骤,而渣油是最复杂的石油组分,即使目前最先进的分析技术还不足以获得其详细的分子组成信息。以现有的分析技术为基础,借助计算机模拟计算是目前渣油分子建模最主要的方法。
目前主要有结构导向集总、分子同系物矩阵以及统计重构3种分子建模的方法。1992年,Quann等[1]提出了结构导向集总的方法,采用22个结构向量来描述烃类分子组成和反应过程,2005年,Jeff等[2]将其扩展到渣油分子的体系。分子同系物矩阵是由Zhang[3]提出的用矩阵的形式来表示石油馏分的方法,设定石油馏分中可能存在的分子结构特征类型,再以碳数划分确定每个矩阵分子,该模型可以粗略地预测轻质馏分的烃族组成。1990年,Neurock等[4]提出了基于随机抽样的统计重构法来模拟石油的分子组成,其根据石油中各个分子特征的分布规律,利用概率密度函数进行抽样,以石油的常规分析数据为基础,生成大量的虚拟分子来表示石油的组成。在此基础上,Verstraete等[5]对模型做了进一步的扩展,增加了分子特征分布的选择,提出了用统计抽样生成虚拟分子,再利用熵值最大化的方法确定分子相对含量,并应用于渣油体系的分子建模中。
本研究从结构导向集总模型出发,选定可以表示渣油的组成特点的65种单核分子和12种多核分子作为种子分子,再以侧链碳数进行详细划分,构建共计3 749个虚拟分子,并采用15种基本的结构向量来表示各个分子的组成。利用概率密度函数来表示渣油中各分子特征的分布规律,确定分子含量的表示方法,结合渣油的基本物性和模拟退火算法计算渣油中分子的相对含量。选取3种渣油进行分子建模研究,分别确定其分子特征分布的参数,计算各渣油的平均物性、分子的摩尔分数和质量分数。
1 渣油分子建模
1.1 分子的构建
提出了以分子同系物矩阵为基本思想的渣油分子建模方法,即假设每个渣油分子都是由核加侧链的形式组成,以分子类型和不同的侧链碳数具体划分。基于渣油的各种分析数据,可以确定渣油中可能存在的分子类型有:链烷烃类、硫醚类、环烷烃类、环烷酸类、芳烃类、噻吩类、吡咯类、吡啶类和代表胶质和沥青质分子的多核类这9类分子,这样就基本上涵盖了渣油中所有可能存在的分子类型。结合FT-ICR MS和FD/TOF MS对渣油中分子结构的解析结果,分析渣油中可能存在的分子结构和种类,经过简化和分类整理,确定了65种单核分子和12种多核分子来作为种子分子,如图1、图2所示。
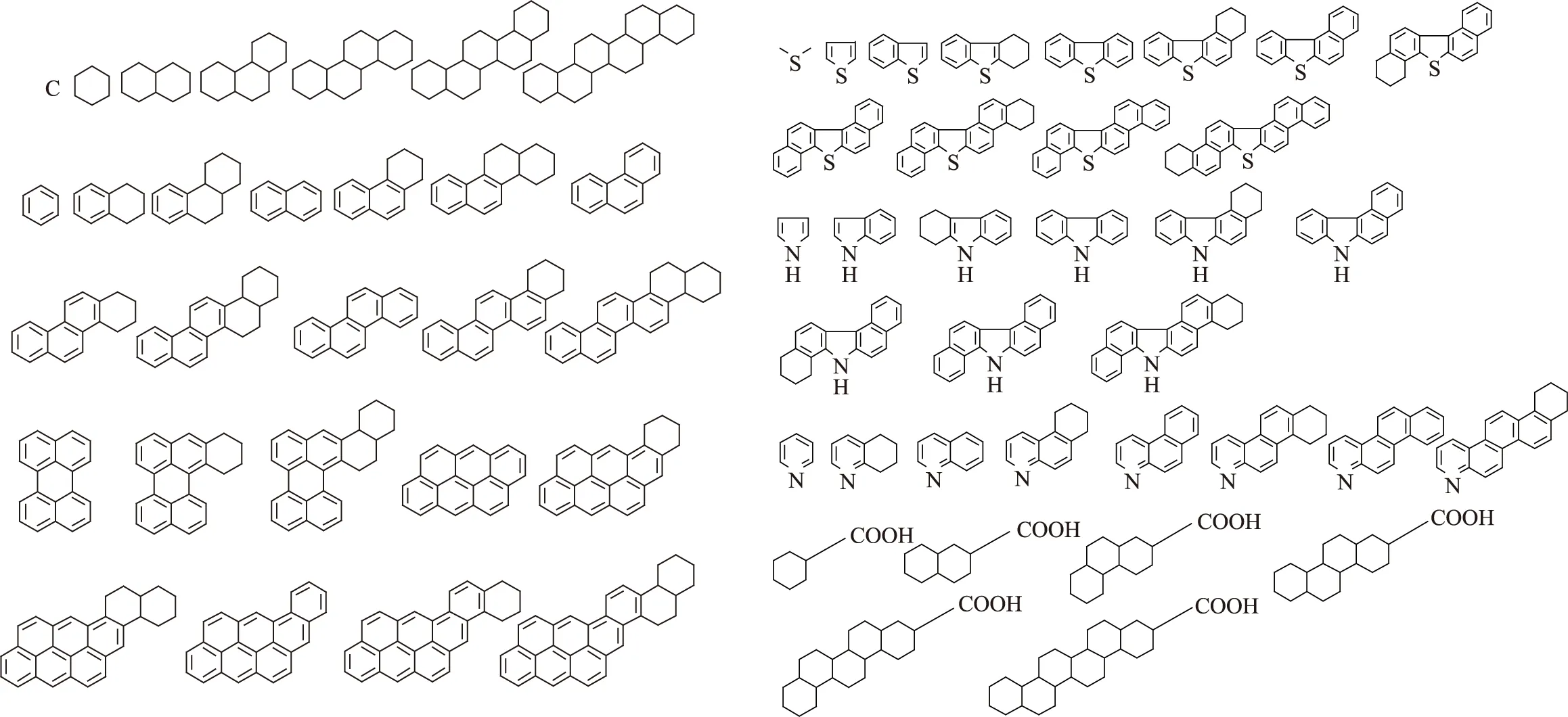
图1 单核种子分子

图2 多核种子分子
按照核加侧链的基本思路,在上述的种子分子分别添加含有1~50个—CH2—的正构烷基侧链。为了方便计算,降低模型的复杂性,分子构建中做出了如下假设:
(1)每个核上只允许连接一个侧链,具体的添加规则是,环烷环优先于芳香环添加,碳原子环优先于杂原子环,外部环优先于内部环。
(2)不考虑烷基侧链的碳链异构形式,即每个碳数侧链的分子都可以表示该碳数下所有可能分子的集总。
(3)多核分子中每个核心上添加的烷基侧链碳数保持一致。
这样共计构建3 749个虚拟分子,相对分子质量为16~5 372,涉及元素C,H,O,N,S,包括链烷烃、硫醚、环烷烃、芳烃、环烷酸、噻吩类、吡咯和吡啶类分子,还有胶质和沥青质分子,这些分子可以代表渣油的分子组成。假设本文中所涉及的渣油都是由上述3 749个分子组成,渣油中分子相对含量的不同导致各类渣油的性质不同。
1.2 分子的矩阵表示
渣油分子是由确定的结构片段组成的,在结构导向集总的22种向量的基础上进行精简和添加,设定了15种基本的结构向量来表示渣油分子,包括A6,A4,A2,N6,N4,N2,S1,S2,N1,N2,R,Me,O,表示多核分子的核间桥链AA,表示分子不饱和度的H,选定的结构向量如图3所示。每个渣油分子都可由确定的分子矩阵来表示,其反应过程和分子物性的计算也可由分子矩阵的计算和变换来表示。通过结构向量的化学计量矩阵可以确定各个分子的原子数以及化学式,表1为各个结构向量的化学计量矩阵。为了避免同一种分子可由不同的结构向量组来表述和不同分子可以由同一结构向量组表示的情况,对分子的矩阵表示做以下规定:
(1) A6优先,即分子中优先出现芳环的表示。
(2) A4优先,即在表示含有多个芳环的分子时首先考虑A4的组成。
(3) N4优先,即在多环的环烷烃分子中,N4优先存在。
(4)H表示芳烃除外的分子不饱和度,链烷烃的H=0,α-烯烃的H=-1,H<-1时呈共轭的形态。
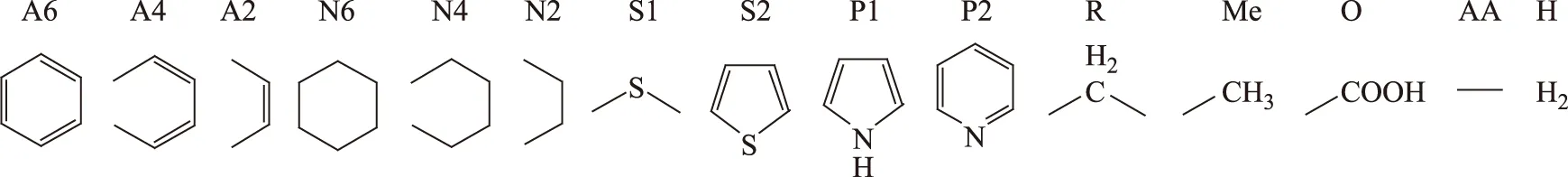
图3 结构向量
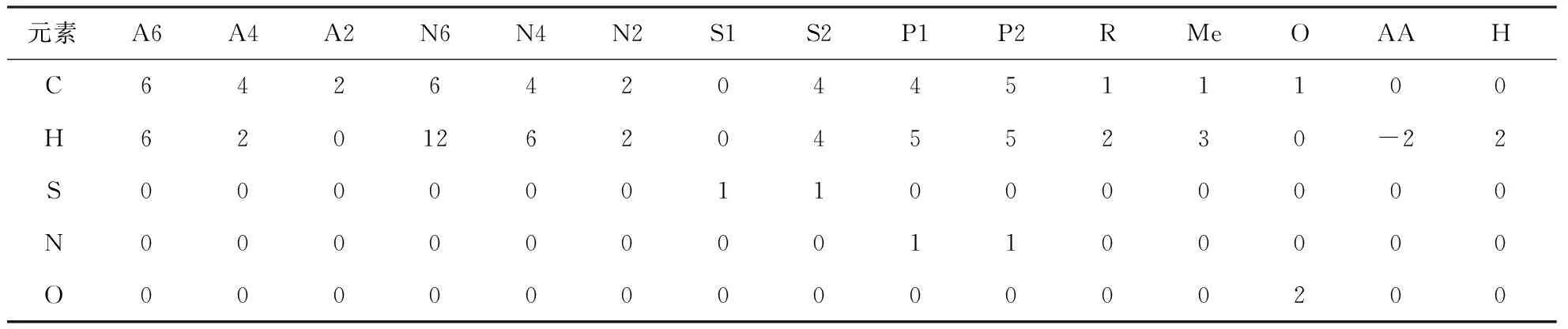
元素A6A4A2N6N4N2S1S2P1P2RMeOAAHC642642044511100H62012620455230-22S000000110000000N000000001100000O000000000000200
2 分子相对含量计算
完成渣油的分子建模还需要确定其中各分子的相对含量,由于渣油中所含分子数较多,以各分子含量为变量进行多目标优化搜索显然是不现实的。祝然[6]对渣油进行窄馏分切割,对各馏分进行GC-MS、元素含量、馏程以及相对密度等分析确定各分子含量的初值、以渣油的整体性质如元素含量、四组分含量、相对密度、相对分子质量等作为目标函数、采用非线性回归的方法计算渣油中的各分子相对含量。该方法计算结果精度高,但是需要实沸点蒸馏、GC-MS等大量的实验分析,计算过程也较为繁琐。由于石油中的分子数目巨大,可以利用统计分布的思想来研究石油中烃类分布的规律。de Oliveira等[7]用指数分布、直方分布和γ分布来表示渣油中各分子特征的分布,利用统计抽样和熵值最大化的方法来对几种不同来源的减压渣油进行分子建模。沈荣民等[8]利用χ2分布来表示各分子特征的分布,通过蒙特卡罗模拟随机抽样来构建延迟焦化原料油的分子。本研究以渣油中各分子特征的分布规律为基础,结合概率密度函数和模拟退火算法来计算各分子的相对含量。
2.1 分子特征分布
由于渣油的物理性质如沸点、相对分子质量等都是由各分子的结构特征综合表现的结果,所以假设渣油中的分子类型、芳环数、环烷环数、多核分子的核心数以及各类分子的侧链碳数分布都可以用γ分布来表示,其概率密度函数由式(1)表示。

(1)
式(1)中χ表示各分子特征的取值,xmin为其最小值,α为形状参数,β为尺度参数。本研究所涉及的9种分子特征分布如表2所示。每个渣油分子都可以通过几种特定的分子特征来表示,如单核的芳烃类分子是由其分子类型、分子中芳环数、环烷环数以及侧链碳数这几种特征分布来确定的,这样用组成分子的各个特征的分布函数的乘积就可以表示分子的摩尔分数,确定各分子特征的分布参数即可计算出每个分子的摩尔分数。

表2 所选的分子特征分布
2.2 模拟退火计算
结合各分子的结构向量矩阵组成和化学计量矩阵可以方便地计算其基本物性,包括平均相对分子质量、元素含量、芳香碳率和环烷碳率等;通过基于结构向量的基团贡献法则可以计算其沸点、密度等。计算出各分子的基本物性,再结合各分子的摩尔分数就可计算出渣油对应的宏观物性。选定渣油的平均相对分子质量M、碳含量C、氢含量H、硫含量S、氮含量N、饱和烃含量Sa、芳烃含量A以及胶质和沥青质含量RA这几种物性,以这些物性的实验值和计算值的相对误差的平方和F来构建目标函数,通过模拟退火算法进行优化搜索,即可确定各分子特征分布的参数,再利用分子含量的表达式即可计算出渣油中各个分子的摩尔分数。图4为分布参数计算的流程。
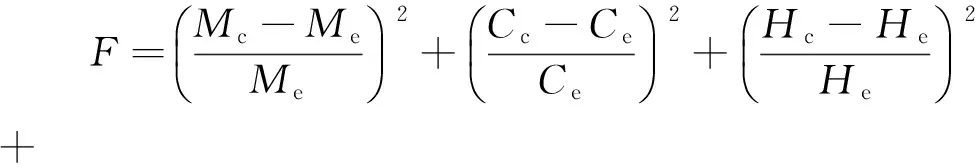
(2)
式(2)中的下角标c表示计算值,e表示实验值。

图4 分布参数计算流程
3 模拟实例
选取3种渣油进行分子建模研究,假设每种渣油都是由上述的3 749个分子组成,各渣油性质的不同主要是由于其中各分子的相对含量不同。利用上述方法分别对这3种渣油进行计算,模拟计算所得的各渣油的分子特征分布的参数如表3所示,表4列出了3种渣油各个物性的实验分析值和计算值的比较。结果表明,模拟计算得到的各物性与实验测定值基本吻合,其中有些物性的相对误差较大,可能是由于其含量较低,所占的权重系数较小,通过增加目标函数中的物性项如密度和沸程分布等可以进一步提高计算精度。
根据各分布参数计算出各分子的摩尔分数以及质量分数,再以不同的相对分子质量进行划分,确定渣油的相对分子质量分布。图5~图7分别表示这3种渣油的芳香分相对分子质量分布。由图5~图7可知,这几种渣油的整体相对分子质量分布都呈现明显的γ分布,符合渣油典型的凝胶色谱的分析结果[9]。其中,每种渣油的分布形状不同,峰值也不同,可以反映出各渣油中芳烃的相对含量不同,渣油3和渣油1中的中芳烃和重芳烃含量较高,而渣油2则是轻芳烃的含量较高。上述计算结果表明,本研究所构建的3 749种分子能够反映渣油的整体特性,分子含量的计算方法是合理的。

表3 3种渣油的各分子特征的分布参数
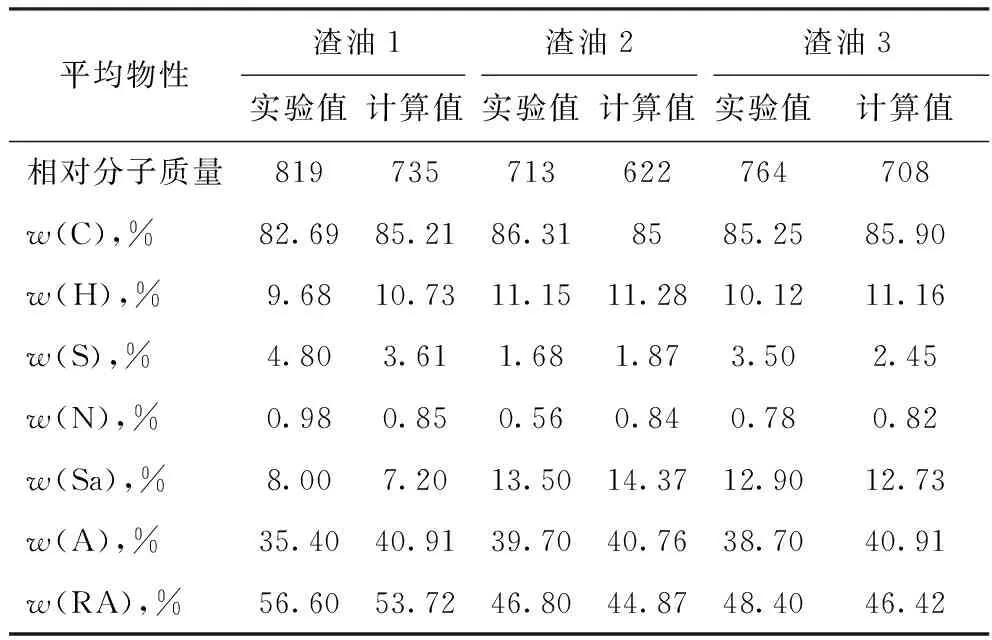
表4 3种渣油的物性实验值和计算值的对比
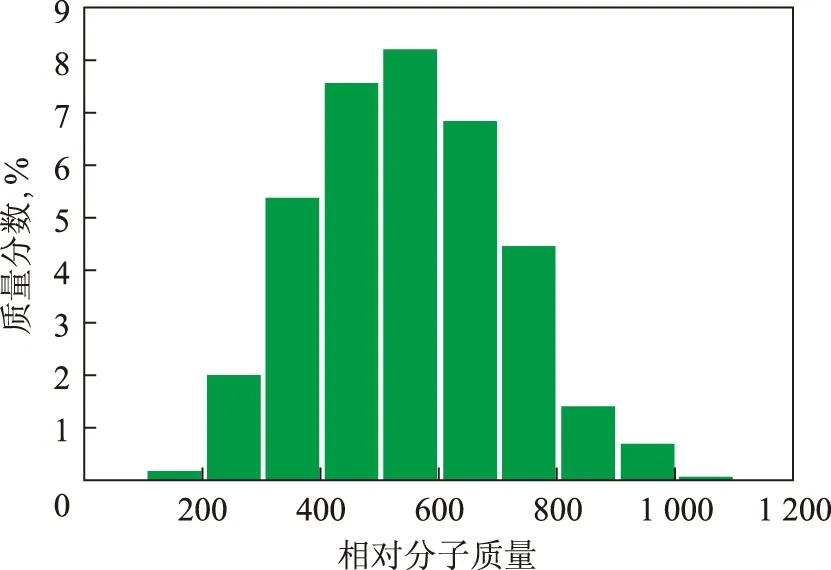
图5 渣油1的芳香分相对分子质量分布

图6 渣油2的芳香分相对分子质量分布
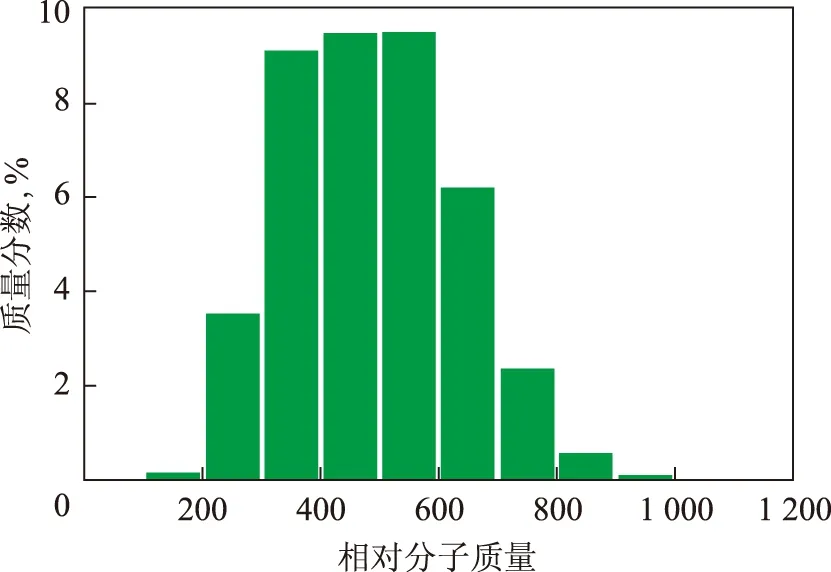
图7 渣油3的芬香分相对分子质量分布
4 结 论
(1) 基于分子同系物矩阵的方法,选定65种单核分子和12种多核分子作为种子分子,以侧链碳数再具体划分,共构建了3 749个分子来表示渣油的分子组成,并假设设计的3种渣油都是由这些分子组成,且各渣油中分子的相对含量不同。
(2) 在结构导向集总模型的基础上进行精简和添加,设定了15种基本的结构向量,用这些结构向量组成的分子矩阵来表示渣油分子,可以用于分子反应过程的表示以及分子物性的计算。
(3) 提出了一种渣油分子摩尔分数的计算方法:利用渣油中各分子特征的分布来确定分子含量的表达式,以渣油几种基本物性建立目标函数,通过模拟退火算法来计算渣油中各分子的含量。
(4) 选取3种渣油进行分子建模研究,模拟计算结果表明,渣油各物性的计算值和实验测定值基本吻合,每种渣油芳香分的相对分子质量也呈现明显的γ分布,这3 749个分子可以基本反映渣油的整体性质,利用分子特征分布来计算渣油分子的相对含量是可行的。
[1] Ruann R J,Jaffe S B.Structure-oriented lumping:Describing the chemistry of complex hydrocarbon mixtures[J].Ind Eng Chem Res,1992,52(31):2483-2497
[2] Jaffe S B,Freund H,Olmstead W N.Extension of structure oriented lumping to vacuum residua[J].Ind Eng Chem Res,2005,44(26):9840-9852
[3] Zhang Pengbin.Molecular modeling of petroleum processes[D].Manchester:University of Manchester Institute of Science and Technology,1999
[4] Neurock M,Nigam A,Libanati C,et al.Monte Carlo simulation of complex reaction systems:Molecular structure and reactivity in modeling heavy oils[J].Chem Eng Sci,1990,45(8):2083-2088
[5] Verstraete J J,Schnongs P,Dulot H,et al.Molecular reconstruction of heavy petroleum residue fractions[J].Chemical Engineering Science,2010,65(1):304-312
[6] 祝然.结构导向集总新方法构建催化裂化动力学模型及其应用研究[D].上海:华东理工大学,2013
[7] de Oliveira L P,Vazquez A T,Verstraete J J,et al.Molecular reconstruction of petroleum fractions:Application to vacuum residues from different origins[J].Energy & Fuels,2013,27(7):3622-3641
[8] 沈荣民,蔡军杰,江洪波,等.延迟焦化原料油分子的蒙特卡罗模拟[J].华东理工大学学报(自然科学版),2005,31(1):56-61
[9] 葛家齐,杨明彪,陆婉珍.五种国产减压渣油的相对分子质量分布[J].石油炼制与化工,1982,13(12):12-16
SIMULATION STUDY OF MOLECULAR COMPOSITION OF RESIDUE
Ren Xiaotian, Yan Long, Shen Haiping
(SINOPECResearchInstituteofPetroleumProcessing,Beijing100083)
Based on the molecular type homologous series matrix, a structure oriented lumping (SOL) method was established to simulate molecular composition of residue, where 65 single cores and 12 multi-cores assumed structure were chosen as the seed molecules which were divided by carbon number of the side chain, resulting in 3 749 kinds of virtual molecules to represent the residue molecular compositions. Each molecule was composed of 15 kinds of basic structure vectors to represent the reaction process of molecules and to calculate the individual properties. With the structural attribute distributions of the residue, the mole fractions of each molecule can be confirmed, and the bulk properties were derived from the calculated mole fractions and the individual properties. Therefore, the mole fractions were determined based on the parameters of the attribute distributions by simulation annealing algorithm. As the results of 3 kinds of residual samples show that the calculated values of bulk properties of residue are in accordance with the measured ones, the distribution of the molecular weight reveals obvious gamma distribution for relative molecular mass of each residue aromatics. It is feasible to represent the residue with the 3 749 kinds virtual molecules, and the method based on the structural attribute distributions can well be used to calculate the mole fractions.
residue; structure vector; molecular matrix; structural attribute distributions; simulated annealing algorithm
2015-05-29。
任小甜,硕士研究生,主要从事重油加工分子模拟的研究工作。
申海平,E-mail:shenhp.ripp@sinopec.com。
猜你喜欢
石油炼制与化工(2023年1期)2023-02-07
能源工程(2022年1期)2022-03-29
韩国语教学与研究(2021年1期)2021-07-29
山东农业大学学报(自然科学版)(2021年3期)2021-07-29
煤气与热力(2021年6期)2021-07-28
石油沥青(2019年4期)2019-09-02
世界农药(2019年2期)2019-07-13
石油炼制与化工(2018年5期)2018-03-23
石油学报(石油加工)(2018年1期)2018-03-05
粘接(2017年4期)2017-04-25
