
制备条件对Ni2P/TiO2-Al2O3 催化剂加氢脱硫性能的影响
2015-07-25 09:11苑丹丹张永江李锋宋华
化工进展 2015年7期
苑丹丹,张永江,李锋,宋华,2
(1 东北石油大学化学化工学院,黑龙江 大庆 163318;2 东北石油大学石油与天然气化工省重点实验室, 黑龙江 大庆 163318)
由于日益严格的环保要求对油品中硫含量的要求以及原油质量的普遍下降,加氢脱硫备受人们关注[1-2]。过渡金属磷化物作为一类加氢精制催化剂新材料,其中晶态Ni2P 不仅活性高,而且具有良好的稳定性,受到人们普遍关注。目前所报道的方法大致可以分为高温制备和低温制备。高温制备主要采用程序升温还原法,低温制备采用液相法、次磷酸盐热分解法等。程序升温还原法的特点是不需要昂贵、剧毒的化学试剂以及高压的反应条件,流程相对简单。络合剂被广泛应用在分子筛的改性和催化剂制备等方面[3-4],陈俊任等[5]发现柠檬酸络合剂能提高NiW 催化剂金属活性组分的分散度,改变金属组分的存在状态,促进高活性的六配位八面体Ni物种的形成,显著改善NiW 催化剂焦化蜡油加氢性能。Wang 等[6]利用柠檬酸与镍的螯合作用,将柠檬酸加到硝酸镍和磷酸氢二铵混合溶液中,干燥、焙烧后采用程序升温还原法制备出了比表面积高达216m2/g 的非负载型HDS Ni2P 催化剂。本文作者课题组[7-9]研究了柠檬酸对负载型Ni2P/TiO2-Al2O3催化剂加氢活性的影响,但催化剂的制备方法和制备条件对催化剂的加氢脱硫活性影响也很大,本文采用溶胶-凝胶法制备TiO2-Al2O3复合载体,以柠檬酸为络合剂,采用浸渍法制备了负载型Ni2P/TiO2- Al2O3催化剂,考察了浸渍方法、Ni/P 摩尔比、Ni2P负载量对催化剂加氢脱硫性能的影响。
1 实验部分
1.1 载体的制备
将一定体积的乙醇缓缓移入钛酸四丁酯中(二者体积比为4∶1),再加入Al2O3粉末,搅拌30min后缓慢滴加配好的乙醇、蒸馏水和冰乙酸的混合溶液(体积比为5∶2∶2),继续搅拌2h,静置陈化12h,120℃烘箱干燥,研磨并筛好,于550℃恒温焙烧3h 即可得TiO2-Al2O3载体粉末,记为TA。
1.2 催化剂的制备
(1)共浸渍法 用计量比的硝酸镍(北京双环化学试剂厂)与NH4H2PO4(西安化学试剂厂)的水溶液浸渍一定质量的TA 和柠檬酸CA 粉末,120℃干燥后,再于500℃焙烧3h,制得不同磷源的Ni2P/TA 催化剂的前体,其标称Ni/P 摩尔比为1∶2、Ni2P 负载量为30%。保持其他条件不变,改变Ni/P摩尔比制得标称Ni/P 摩尔比分别为3∶1、2∶1、1∶1、1∶2、1∶3 的Ni2P/TA 催化剂前体;标称Ni/P摩尔比1∶1 等其他条件不变,改变Ni2P 负载量制得Ni2P 负载量分别为10%、20%、30%、40%的Ni2P/TA 催化剂前体。反应前将催化剂前体样品用氢气(200mL/min)还原,先以3℃/min 升至350℃,再以1℃/min 升至700℃还原2h,然后降至室温,切换30mL/min N2钝化处理2h。
(2)分步浸渍法 保持标称Ni/P 摩尔比1∶2和Ni2P 质量负载量30%等其他条件不变,用计量比的硝酸镍和NH4H2PO4采用分步浸渍法制备。
1.3 催化剂的表征
催化剂的X 射线衍射(XRD)分析在日本理学公司D/max-2200PC 型X 射线衍射仪上进行,采用Cu Kα 辐射,管电压40kV,管电流30mA,扫描速率10°/min,扫描范围10°~80°。催化剂比表面积是在NOVA2000e 测定仪上,利用低温(-196℃)氮气吸附法测定。样品首先在100℃、1.3kPa 预处理4h。
1.4 催化剂的评价
催化剂评价在固定床高压微反装置中进行。反应前催化剂在500℃通氢气(80mL/min)处理2h,再降至反应温度,测定其HDS 活性。采用的模型化合物为二苯并噻吩、十二烷烃和环己烷的混合溶液,其体积分数分别为2%、1%和97%。其中,环己烷为溶剂,十二烷为内标物。反应条件均为360℃、3.0MPa、氢油比500 (体积比)、空速2.0h-1。得到的液体产物分析在日本岛津公司的GC-14C 型气相色谱仪上进行。
2 结果与讨论
2.1 催化剂的XRD 物相分析
2.1.1 不同浸渍方法制备的催化剂XRD 分析
图1 为不同浸渍方法的催化剂的XRD 谱图。由图1 可知,不同浸渍方法的催化剂中TiO2均以锐钛矿型存在,催化剂经还原后得到了不同的活性相磷化镍;先浸镍和先浸磷时,在2θ 约为38.4°、41.7°、44.4°、46.9°、48.9°出现明显的衍射峰,这与Ni12P5的主要衍射峰一致,说明主要还原产物为Ni12P5而不是Ni2P;当共浸渍时,样品在2θ 约为 40.7°、44.6°、47.3°和54.1°处出现明显的衍射峰,这与Ni2P 相的主要衍射峰一致,说明此时样品还原后主要物相均为Ni2P。以上结果表明,采用不同浸渍方法能够改变Ni2P/TA 催化剂活性相的种类,适宜的浸渍方法是共浸渍。

图1 不同浸渍方法制备的催化剂的XRD 谱图
2.1.2 不同Ni/P 摩尔比的催化剂的XRD 分析
图2 为不同Ni/P 摩尔比的催化剂的XRD 谱图。由图2 可知,不同Ni/P 摩尔比的催化剂中TiO2均以锐钛矿型存在,催化剂经还原后得到了不同的活性相磷化镍。当Ni/P 摩尔比低于1∶1 时,样品在2θ 约为40.7°、44.6°、47.3°和54.1°处出现明显的衍射峰,这与Ni2P 相的主要衍射峰一致,说明催化剂中活性相磷化镍均为Ni2P;Ni/P 比为1∶3时,相应的Ni2P 的衍射峰较弱,且随Ni/P 摩尔比的提高,衍射峰强度增强;当Ni/P 摩尔比为2∶1时,形成的物相除了Ni2P 外还有一部分Ni3P;当Ni/P 比为3∶1 时,形成大量的Ni3P 物相,表明镍含量过高时,会导致由亚稳相Ni2P 转变为稳定相Ni3P[10]。
2.1.3 不同Ni2P 负载量的催化剂的XRD 分析

图2 不同Ni/P 摩尔比的催化剂的XRD 谱图

图3 不同Ni2P 负载量的催化剂的XRD 谱图
图3 为不同Ni2P 负载量的催化剂的XRD 谱图。 由图3 可知,样品中TiO2均以锐钛矿型存在,各催 化剂样品在2θ 约为40.7°、44.6°、47.3°和 54.1°处出现明显的衍射峰,这与Ni2P 相的主要衍射峰一致,表明催化剂的磷化镍相主要为Ni2P。Ni2P负载量为10%时,形成的Ni2P 衍射峰强度较弱,随着Ni2P 负载量的提高,Ni2P 的衍射峰强度逐渐 增强。
2.2 催化剂的BET 分析
表1 为不同Ni/P 摩尔比和负载量的催化剂的BET 分析结果。由表1 可知,不同Ni/P 摩尔比的催化剂的孔径随摩尔比的降低,先增加后减小。当Ni/P摩尔比从3∶1 减小至1∶1 时,催化剂的比表面积略微减小,变化幅度不大;Ni/P 摩尔比从1∶1 继续减小时,催化剂的比表面积增大且增幅较大;当Ni/P 比为1∶2、1∶3 时,催化剂的比表面积接近200m2/g,与TA 复合载体的比表面积相差不大,表明当Ni/P 比为1∶2、1∶3 时,复合载体上磷化物(活性组分Ni2P)的生成量较少,不利于HDS 活性的提高。结果表明,加入CA 后不同Ni/P 摩尔比的催化剂比表面积均比Ni2P/TA 催化剂[11]有较大提高,形成的物相也有差别(见XRD 分析)。Ni/P 摩尔比过高或者过低都不利于活性相Ni2P 的形成,Ni/P 比为1∶1 时,催化剂的比表面积为126.75m2/g且形成的活性相为单一的Ni2P 物相。不同Ni2P 负载量的催化剂的孔径随负载量的增加而减小。当Ni2P 负载量增加时,催化剂的比表面积先增大后减少。Ni2P 负载量30%时比表面积达到最大值,为126.75m2/g。在一定范围内催化剂的比表面积越大,越有利于气固非均相催化反应的进行。当Ni2P 负载量超过40%时,催化剂比表面积下降,不利于催化反应的进行,这表明适宜的Ni2P 负载量为30%。

表1 不同Ni/P 摩尔比和负载量的催化剂的比表面积
2.3 催化剂的催化反应性能
2.3.1 浸渍方法对催化剂催化性能的影响
图4 为不同浸渍方法制备的催化剂对二苯并噻吩的HDS 活性图。由图4 可知,在相同反应条件下进行HDS 反应时,共浸渍法制备的催化剂活性最好,最高转化率在反应4h 时达到99.5%。反应时间在2h 之前,不同催化剂样品DBT 转化率均随反应时间的增长而增大;但在2h 之后,先浸渍P 的催化剂和先浸渍Ni 的催化剂反应活性却随反应时间的继续增长而呈下降趋势。这主要是因为浸渍顺序的不同,使得Ni、P 在载体上的负载情况发生了变化,由XRD 分析可知,先浸渍P 的催化剂和先浸渍Ni 的催化剂的活性相Ni2P 较少,形成了大量的Ni12P5,表明采用不同浸渍方法能够改变Ni2P/TA 催化剂活性相的种类,以致影响其HDS 活性。
2.3.2 Ni/P 摩尔比对催化剂催化性能的影响
图5 是不同Ni/P 摩尔比催化剂对二苯并噻吩HDS 活性图。从图5 可知,在反应从1h 进行到2h这段时间内,不同Ni/P 比的催化剂的HDS 转化率均随反应时间的增加而增加;在反应从2h 进行到5h 这段时间内,Ni/P 比为1∶1 和1∶2 的催化剂HDS 转化率曲线基本趋于平稳,Ni/P 比为1∶3、2∶1 和3∶1 的催化剂HDS 转化率曲线随反应时间明 显呈下降趋势。当Ni/P 比为1∶1 时,催化剂的HDS转化率随着时间的增加而先增加后减少,当反应时间为4h 时,催化剂的HDS 转化率达到最大;而当Ni/P 比为1∶2 时,在反应时间3h 后催化剂的HDS转化率随着反应时间增加而减少。结果表明,当Ni/P比为1∶1 时,可获得较高的二苯并噻吩HDS 的转化率,与XRD 和BET 表征结果相吻合。

图4 浸渍方法对制备的催化剂对二苯并噻吩HDS 活性的影响

图5 Ni/P 摩尔比催化剂对二苯并噻吩HDS 活性的影响
2.3.3 Ni2P 负载量对催化剂催化性能的影响
图6 是不同Ni2P 负载量催化剂对二苯并噻吩的HDS 活性图。由图6 可知,在反应从1h 进行到2h这段时间内,不同Ni2P 负载量催化剂的HDS 转化率均随反应时间的增加而增加。在反应从2h 进行到5h 这段时间内,Ni2P 负载量为30%的催化剂HDS转化率随反应时间趋于稳定,当反应时间为4h 时转化率达到最大,为99.5%;而Ni2P 负载量为10%的催化剂的HDS 转化率随时间的增加而先增加后减少,反应4h 后迅速降低;负载量为20%和40%的催化剂HDS 转化率随反应时间增加呈下降趋势。当Ni2P 负载量为40%时,转化率较Ni2P 负载量为30%的催化剂低,其原因可能是Ni2P 负载量为40%时活性组分在载体上的分散性较差,比表面积减少(表2),不利于HDS 活性的提高。
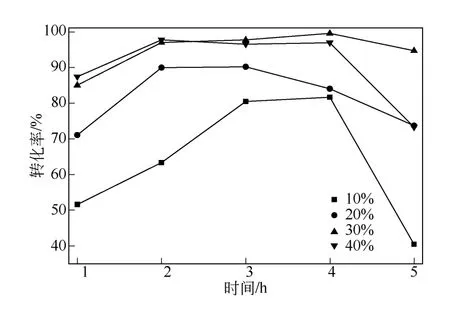
图6 Ni2P 负载量对催化剂对二苯并噻吩HDS 活性的影响
3 结 论
采用溶胶-凝胶法制备了TiO2-Al2O3复合氧化物,并以柠檬酸为络合剂采用浸渍法制备负载型Ni2P/TiO2-Al2O3催化剂前体,然后程序升温H2还原法制备催化剂。实验结果表明,在Ni/P 比从1∶3增大到3∶1 的过程中,Ni2P 的衍射峰先增强后减弱。当Ni/P 比为1∶1 时,得到的Ni2P 衍射峰最强,继续增加Ni/P 摩尔比到3∶1 时,会导致由亚稳相Ni2P 转变为稳定相Ni3P。采用共浸渍方式制备的样品的物相为Ni2P,且衍射峰强度较大,分步浸渍催化剂样品形成的物相为Ni12P5,表明适宜的制备方法是共浸渍法。Ni2P 的衍射峰随Ni2P 负载量的增加先增强后不变,当Ni2P 负载量为30%时,活性相为 Ni2P 且此时的Ni2P 衍射峰较强,适宜的Ni2P负载量为30%。
[1] Oyama S T,Gott T,Zhao H Y,et al.Transition metal phosphide hydroprocessing catalysts:A review[J]. Catalysis Today,2009,143:94-107.
[2] 仝建波,蔺阳,刘淑玲,等. 加氢脱硫催化剂载体的研究进展[J]. 化 工进展,2014,33(5):1170-1179.
[3] Ge H,Wen X D,Ramos M A,ea tl. Carbonization of ethylenediamine coimpregnated CoMo/Al2O3catalysts sulfided by organic sulfiding agent[J]. ACS Catal.,2014,4(8):2556–2565.
[4] Yoshimura Y,Sato T,Shimada H,et al. Preparation of nickel-tungstate catalysts by a novel impregnation method[J]. Catalysis Today,1996,29:221-228.
[5] 陈俊任,周亚松. 柠檬酸对Ni-W 焦化蜡油加氢处理催化剂性能的影响[J]. 化工学报,2007,58(9): 2244-2248.
[6] Wang R , Smith K J. Hydrodesulfurization of 4,6-dimethyldibenzothiophene over high surface area metal phosphides[J]. Applied Catalysis A:General,2009,361:18-25.
[7] 宋华,张永江,宋华林,等. 柠檬酸对Ni2P/TiO2-Al2O3催化剂加氢脱硫性能的影响[J].物理化学学报,2012,28(3):661-666.
[8] Song H,Wang J,Wang Z D,et al. Effect of titanium content on dibenzothiophene HDS performance over Ni2P/Ti-MCM-41 catalyst[J]. Journal Catalysis,2014,331:257-265
[9] Song H,Dai M,Song H L,et al. A solution–phase synthesis of supported Ni2P catalysts with high activity for hydrodesulfurization of dibenzothiophene[J]. Journal of Molecular Catalysis A:Chemical,2014,385:149-159.
[10] 宋华,郭云涛,李锋,等. Ni2P/TiO2-Al2O3催化剂的制备及其加氢脱硫、脱氮性能[J]. 物理化学学报,2010,26 (9):2461-2467
[11] 宋华,于洪坤,武显春,等. TiO2-Al2O3载体的制备及Ni2P/TiO2-Al2O3催化剂上的同时加氢脱硫和加氢脱氮反应[J]. 催化学报,2010,31(4):447-453.
猜你喜欢
化学工程师(2023年1期)2023-02-17
农业研究与应用(2021年2期)2021-08-12
理化检验-化学分册(2020年12期)2021-01-26
世界有色金属(2020年4期)2020-05-16
上海农业科技(2019年1期)2019-02-22
中国果业信息(2018年5期)2018-01-17
当代化工研究(2016年1期)2016-03-16
合成化学(2015年10期)2016-01-17
湖南大学学报·自然科学版(2014年7期)2014-11-28
应用化工(2014年9期)2014-08-10
