
阿尔茨海默病的可能药物靶点和临床治疗研究进展
2015-07-24 19:02毕丹蕾文朗熊伟申勇
中国药理学与毒理学杂志 2015年4期
毕丹蕾,文朗,熊伟,申勇
(中国科学技术大学生命科学学院,生命的化学基础协同创新中心,中国科学院脑功能及脑疾病重点实验室,1.神经退行性疾病研究中心,2.感知觉、情绪与认知综合神经科学实验室,安徽合肥230027)
阿尔茨海默病的可能药物靶点和临床治疗研究进展
毕丹蕾1,文朗1,熊伟2,申勇1
(中国科学技术大学生命科学学院,生命的化学基础协同创新中心,中国科学院脑功能及脑疾病重点实验室,1.神经退行性疾病研究中心,2.感知觉、情绪与认知综合神经科学实验室,安徽合肥230027)
编者按:阿尔茨海默病是导致老年痴呆的主要神经退行性疾病,在65岁以上人群的发病率高达7%~10%,然而尚无有效的治疗手段。随着我国老龄化进程的加快,尤其是在“中国脑计划”即将启动的大背景下,阿尔茨海默病研究的意义越发凸显。基于此,本刊邀请阿尔茨海默病研究领域专家申勇等教授撰文,介绍国际上关于阿尔茨海默病可能药物靶点的机制研究和潜在药物的最新临床试验的最新进展。这不仅可以为国内阿尔茨海默病领域的科研工作者提供参考,也可以为对发展阿尔茨海默病药物有兴趣的医师、药审专家、政策制定人,甚至投资者等提供有效资讯,通过各相关领域专家的共同努力,加速阿尔茨海默病临床药物的研发。
阿尔茨海默病(AD)是高发于65岁以上人群的神经退行性疾病,其病理特征是大脑中β淀粉样蛋白(Aβ)聚集形成的老年斑、过度磷酸化的tau蛋白聚集而成的神经元纤维缠结、长期炎症反应以及神经元死亡等。因此,除了衰老之外,Aβ聚集、tau过度磷酸化、慢性炎症及神经元死亡被认为是AD的主要发病假说之一。本文介绍了基于以上假说的AD治疗靶点的研究及药物研发进展。目前进展最快的药物都基于神经保护假说,全部5个小分子AD药物都是通过调节兴奋性神经递质传递通路而改善AD患者认知障碍的,但是它们的疗效非常有限。基于这一机制的新药研发也主要集中在抑制兴奋性递质受体这一方向,然而进展有限。基于tau假说的药物主要是通过抑制tau的磷酸化、异常聚集及病理扩散。然而,由于无法特异性抑制tau的磷酸化,目前进展较快的是tau疫苗。目前处于临床试验中的4个tau相关药物有2个是tau的主动免疫疫苗,均处于Ⅰ期临床中。在Aβ假说方面,药物研发思路主要集中在抑制Aβ合成/聚集、促进Aβ清除上。由于
申勇,博士,中国科学技术大学教授,博士生导师。国家千人学者获得者,教育部长江特聘讲座教授。在南京大学生物学系获学士学位,中国科学院上海生理学研究所获硕士学位。美国纽约州立大学获神经科学博士学位。曾任美国Abbott(亚培)制药公司任脑疾病药物靶点研发小组组长及资深神经分子药理学研究员,参与制定和发展治疗老年退行性疾病药物。之后受邀作为罗伯特老年痴呆病研究中心主任,资深研究员及教授在亚利桑那州太阳城Sun Health Research Institute工作多年,他的团队参与了神经免疫相关药物在老年痴呆病的作用,从事了一系列基础和临床以及药物研发等方面等转化工作。2006年兼任美国Sun Health亚太医学研究中心主任。2010年加盟于佛罗里达州的罗斯坎普医学研究所担任脑疾病及治疗战略研究及治疗中心主任,资深科学家及佛罗里达大学医学院神经病学教授继续从事神经疾病的研究。主要研究方向包括神经退行性病、帕金森病及相关脑血管病的基础及转化研究,包括寻找脑疾病的新药的开发以及在细胞及动物模型上进行化学以及天然产物药物筛选。并牵头美国,欧盟多中心寻找并确定临床早期诊断老年神经退行病标记物和参与相关药物临床实验。共发表100余篇包括Nature Medicine,Journal of Cell Biology,JAMA Psychiatry, Neuron,Proceedings of the National Academy of Science USA, Brain,Neurology,Journal of Neuroscience,Journal of Pharmacology and Experimental Therapeutics,Molecular Pharmacology,American Journal of Medicinal Chemistry等国际学术期刊。申勇教授还参与美国食品与药物监督局以及美国临床前脑疾病方面的药物评审,他获得多项奖项及荣誉称号包括:美国阿尔茨海默病研究协会的“先锋奖(Zenith Award)”以及“老年退行病杰出贡献奖(Outstanding Contributor to Alzheimer Research)”,以及2009年中国教育部颁发的“长江学者奖励计划特聘讲座教授”。2014年入选中组部“千人计划”并于同年回国,任中国科技大学教授及神经退行性疾病研究中心主任。γ-分泌酶抑制剂的临床试验因严重不良反应而失败,目前的热点是β-分泌酶(BACE1)抑制剂以及Aβ疫苗。另外,通过抗炎药物抑制患者脑内的长期慢性炎症也被认为是AD治疗的可能手段之一。虽然,非甾体类抗炎药物尚无成功案例,但仍有以其他炎症因子为靶点的药物,如沙利度胺,处于临床试验中,进展良好,值得期待。总之,目前AD药物研发的主要障碍是药物缺乏临床应用指标,特异性不强,而临床试验多由于不良反应以及疗效不足等原因而失败。虽然如此,目前仍有82种AD药物处于临床阶段,其中18种进入了Ⅲ/Ⅳ期临床。同时,计算机设计靶向药物及CRISPR/Cas9等新技术的发展为AD的治疗带来了希望。
神经退行性疾病;阿尔茨海默病;药物发现;治疗应用;淀粉样β肽类;tau蛋白;炎症
阿尔茨海默病(Alzheimer disease,AD),俗称老年痴呆症,最初是在1906年由德国精神科医师Alois Alzheimer博士对一名多年受到记忆问题、意识模糊以及语言障碍困扰的女性患者的诊断。此后100年来的临床试验研究表明,AD是一种多发于65岁以上人群的神经退行性疾病,临床表现为进行性的记忆损伤、认知障碍、精神症状以及日常生活能力的丧失。目前,由AD导致的痴呆占总痴呆病例的50%~75%[1]。由于AD患者日常行为能力严重受损,不仅给患者家庭带来了情感和经济上的强烈冲击,也给政府财政造成了沉重负担。2010年在国际AD大会上发布的《痴呆病对全球经济的影响》报告显示,仅2010年1年,AD造成的经济损失已达6040万美元,而预计至2050年,AD患者人数将会超过1亿,即现在的3倍[2],这必然导致经济损失的相应增加。因此,找到安全可靠的AD预防及有效治疗手段是目前的迫切需求。大量流行病学研究表明,除了基因以外,导致AD最主要的因素就是老化;其次,大脑损伤、低教育程度、高脂血症、高血压、糖尿病和肥胖等都是风险因子[3-6]。
Alzheimer博士的报告中描述的聚集在神经元周围的淀粉样斑块和神经元内的纤维状物质至今仍被认为是AD的经典病理特征。淀粉样斑也称老年斑,被证明是由β淀粉样蛋白(amyloid beta-protein,Aβ)聚集形成的[7-8];而神经元纤维缠结是由过度磷酸化的tau蛋白聚集而成的[9]。此外,AD还伴随着大脑中的长期炎症反应、氧化应激、突触丧失及功能失调、神经元死亡和大脑萎缩等现象[10-13]。
关于AD发病机制,普遍认为衰老是最大的诱因之一。在65岁之后,罹患AD的概率升高1倍,而当年龄超过85岁时,患病概率高达将近50%[14]。目前,除了衰老之外,AD的发病机制主要有4大假说:基于淀粉样斑块的Aβ假说、基于神经元纤维缠结的tau假说、基于长期炎症反应造成脑损伤的炎症假说和基于突触功能失调及神经元死亡的神经保护假说。目前成功完成临床试验并被批准用于AD的临床治疗的药物全部都是基于神经保护假说的,也就是说,都是神经递质产生或神经递质受体的激动剂或拮抗剂,而基于其他假说研发的药物尚处于基础研究及早期临床试验阶段,前景尚不明朗。本文就AD的4大假说以及基于相应假说所开展的临床试验的研究进展进行介绍。
1 神经保护假说
由于AD患者大脑中出现病理特征的主要区域是颞叶、海马、内嗅皮质、杏仁核以及内侧隔核,这些区域与学习记忆能力密切相关[15]。尽管在AD晚期,突触丢失情况与记忆丧失程度成正比[16],然而,在AD早期,患者无法产生新的记忆,然而此时并未出现中晚期患者大脑中常见的病理改变,比如突触丢失、淀粉样斑块、神经元纤维缠结和神经元死亡等[17]。因此,Selkoe[16]提出Aβ会导致神经突触的功能异常,最终导致了突触丢失,这可能是AD的诱因之一。虽然Aβ聚集导致的淀粉样斑块沉积是AD的主要病理现象,然而,进一步的研究表明,由β分泌酶和γ分泌酶剪切淀粉样前体蛋白(amyloid precursor protein,APP)产生的可溶性Aβ寡聚体或微型Aβ聚积具有神经毒性[18]。它们可能在AD早期,即淀粉样斑块、神经元纤维缠结和神经元死亡出现之前,通过引起兴奋性和抑制性的神经传递系统的失衡而导致导致突触功能的失调。因此,在AD早期,通过药物重新协调兴奋/抑制的平衡有可能在AD发病早期延缓病程进展。事实上,目前被美国食品药物管理局(FDA)批准用于AD临床治疗的全部5个药物都是通过调节兴奋性神经递质的传递,改善AD患者的认知。这其中涉及到的药物靶点包括胆碱能系统和谷氨酸能系统。
1.1 胆碱能传递
胆碱能受体主要有毒蕈碱受体和烟碱型受体。大量证据表明,这两类受体广泛分布于大脑中,尤其是与学习记忆紧密相关的前额叶皮质与海马脑区的毒蕈碱类受体可以缓解Aβ导致的神经毒性作用[19-21]。体外实验表明,脑皮质区毒蕈碱样乙酰胆碱受体M1亚型的激活可显著降低Aβ导致的动作电位发放和兴奋性突触后电流,从而中和Aβ导致的兴奋性神经毒性[20]。Gu等[20]认为,毒蕈碱样乙酰胆碱受体M1亚型的激活可增强γ-氨基丁酸(γamino butyric acid,GABA)能抑制性递质的传递,平衡Aβ引起的兴奋性突触后电流。另一项体外实验表明,毒蕈碱样乙酰胆碱受体M1亚型可促进APP被α-水解酶水解,抑制其被β-与γ-水解酶水解产生具有神经毒性的Aβ[21]。同时,M1亚型的特异性激动剂AF267B可有效缓解AD模型小鼠3XTg的AD样病理以及认知障碍[22-23]。因此,毒蕈碱样乙酰胆碱受体被认为是治疗AD的靶点之一。除了使用激动剂激活受体之外,增加乙酰胆碱的浓度也是干预策略之一。目前,已被FDA批准用于AD治疗的5个小分子药物中,有4个是通过抑制乙酰胆碱酯酶的活性、增加乙酰胆碱的浓度,从而增强胆碱能系统的活性来改善Aβ导致的神经毒性作用的。它们分别是他克林(tacrine)、多奈哌齐(donepezil)、加兰他敏(galantamine)和利凡斯的明(rivastigmine)(表1)。其中,他克林是第一个被批准用于AD临床治疗的可逆性乙酰胆碱酯酶抑制剂,然而由于它具有肝毒性,目前已经基本停用。
多奈哌齐,一种可逆性乙酰胆碱酯酶抑制剂,已经被超过90个国家批准用于轻度及中度AD的临床治疗。它通过增强乙酰胆碱的突触传递延缓AD患者认知障碍的恶化。全球接近230个临床试验表明,短期(半年以内)及长期(长达3年)多奈哌齐干预对轻度和中度AD患者有一定的改善认知的作用,同时,对日常活动障碍和行为症状也有轻度的缓解[24-27];同时,多奈哌齐也可以延缓病程进展,如在美国与加拿大进行的3项,共有超过1800位受试者参加的临床试验中,多奈哌齐可以推迟痴呆大约1年,但是它并不改善轻微认知障碍的指标[28]。同时,它的耐受性良好,最常见的不良反应是胃肠道反应,主要包括腹泻、恶心和呕吐等,也有失眠、疲劳以及尿失禁等,这也是胆碱能药物的普遍不良反应。
另外一个以胆碱能系统为靶点的AD临床药物加兰他敏,它不但是乙酰胆碱酯酶抑制剂,还是尼古丁和毒蕈碱类乙酰胆碱受体变构性的增强剂。一项为期1年的多中心组间平行的随机临床比较了加兰他敏和多奈哌齐对认知、日常生活活动能力、照顾者负担水平以及安全性的影响。结果表明,加兰他敏对认知的改善以及护理负担的减轻都显著优于多奈哌齐,而二者对患者日常活动能力的改善与安全性方面,无显著差异[29]。
利凡斯的明是可逆的乙酰胆碱酯酶和丁酰胆碱酯酶抑制剂,它被广泛用于从轻度到重度AD的临床治疗,也用于帕金森病导致的痴呆。临床试验表明,短期及长期服用利凡斯的明与协同加兰他敏和多奈哌齐有相似的改善轻度及重度AD患者的认知功能,然而对它们的横向对比较少。一项为期2年涉及到994例中度至重度AD患者的临床试验比较了利凡斯的明与多奈哌齐二者的效果与耐受性。试验中,几乎一半的受试者因胃肠道不良反应而退出,其余接受利凡斯的明与多奈哌齐的患者出现了相似的认知和行为改善,而利凡斯的明对日常生活能力和整体功能的改善略优于多奈哌齐[30]。另外,除了偶发的皮肤反应和与使用乙酰胆碱酯酶抑制剂造成的常见不良反应,比如恶心、呕吐、失眠和疲劳等,利凡斯的明导致的胃肠道不良反应显著少于其他药物,因此可以使用较大剂量[31]。此外,还有研究表明,利凡斯的明对心血管因素导致的痴呆可能有缓解作用,治疗血管性痴呆的作用;对患有心血管疾病,比如高血压的AD患者疗效好于“纯”AD患者[32-34]。
1.2 谷氨酸能传递
除乙酰胆碱系统,Aβ还通过影响谷氨酸能系统影响神经元的兴奋性,其影响机制存在多个不同假说,比如小胶质细胞对谷氨酸的回收被阻断、谷氨酸突触传递异常、N-甲基-D-天冬氨酸(N-methyl-D-aspartate,NMDA)受体与AMPA/Kainate受体内吞过程的改变、细胞内Ca2+浓度升高导致的兴奋性毒性等[35]。其中,大量证据支持Aβ通过谷氨酸能受体造成神经毒性。比如,NMDA受体抑制剂MK801可以抵抗Aβ造成的神经毒性,起到神经保护的作用。小分子NMDA受体拮抗剂美金刚于2003年被FDA批准用于AD治疗。目前为止,它是唯一被批准适用于AD治疗的非乙酰胆碱酯酶抑制剂。一项为期28周、涉及252例中重度AD患者的Ⅲ期临床试验证明美金刚可以在一定程度上改善中重度AD患者注意力、整体功能、独立性以及行为能力,后续的涉及175例患者的开放式试验表明,美金刚的临床疗效可以持续1年[36-37]。然而,它对轻度到中度AD患者的疗效仍有争议,临床试验结果也不尽相同。比如,在美国和欧洲分别进行了2项为期6个月、针对轻度到中度AD的临床试验。在美国开展的涉及到403例患者的试验表明,美金刚的疗效略优于安慰剂,在欧洲开展的涉及到470例患者的试验表明,美金刚在试验中期时疗效略优于安慰剂,但试验结束时,美金刚与安慰剂之间并无统计学差异[38-39]。尽管美金刚对早期AD的疗效有争议,也从未被批准用于治疗轻度AD,但临床经常用作早期AD的治疗药物。在临床上,美金刚还通常与乙酰胆碱酯酶抑制剂加兰他敏、多奈哌齐或利凡斯的明被同时使用[40]。一项为期6个月、涉及404例中重度AD患者的临床试验证明,美金刚可以增强已接受多奈哌齐治疗的患者的认知功能及行为改善[41]。
1.3 GABA能传递
目前,以谷氨酸和胆碱能系统为靶点的治疗药物疗效与进展都比较有限。认知障碍的产生被认为与不同类型神经元的结构及功能改变和对神经元退行的敏感程度有关。比如,在海马区,乙酰胆碱能神经元与谷氨酸能神经元对神经毒性最为敏感,而GABA能神经元在AD患者大脑中保留较好,对神经退行具有一定的抵抗能力[42-43]。此外,使用具有神经毒性的Aβ42处理神经元末梢之后,谷氨酸能的海马神经元末梢数目减少,而GABA能无显著改变[44],表明GABA能神经元可能参与了抵抗Aβ造成的AD样病理改变。因此,目前AD研究热点主要集中在调节抑制性的GABA能系统。GABA能系统的激活可以抑制神经元的兴奋性,对防止神经元过度兴奋、维持神经网络稳定具有重要作用。尤其是它对神经元产生θ及γ活动时所需的节律同步有关键作用,因此,对神经元之间的联系及记忆产生有重要意义[45-46]。在表达人源淀粉样前体蛋白(human APP,hAPP)的AD模型小鼠大脑中,Aβ显著增加神经元兴奋性,导致非惊厥类癫痫的发生,同时伴随GABA能神经元出芽、突触抑制增强以及神经可塑性受损[47],提示GABA能系统的激活在补偿Aβ导致的兴奋性神经毒性方面的作用。脑活素(Cerebrolysin)(表1),一种GABAB受体激动剂,在多个临床试验中被证明基本安全并且可以改善轻微到重度AD患者的认知表现[48-49]。但是,小鼠、大鼠及猕猴的实验结果都表明,诺华公司开发的GABAB受体抑制剂SGS 742可以显著改善认知,临床双盲、安慰剂对照的Ⅱ期临床试验证明,它可以显著改善轻度AD患者的认知以及工作记忆[50]。其作用机制可能是通过抑制突触前的GABAB受体,增加谷氨酸和天冬氨酸的释放,诱发突触后去抑制现象从而促进长时程增强(long term pertentiation,LTP)的形成,最终帮助记忆形成[50]。然而SGS 742的Ⅱb期临床试验随后被终止,具体原因不明。脑活素与SGS 742的实验结果表明,增强或抑制GABAB受体都可以改善认知,这可能是因为GABA能也是中枢神经系统的兴奋/抑制平衡中重要的一环,它的再平衡有助于缓解Aβ导致的兴奋/抑制失衡。然而,迄今尚无以GABA能系统为靶点的药物通过临床试验,表明GABA能系统在AD中的作用机制需要进一步研究。另外,考虑到GABA能系统在中枢神经系统中的广泛分布与关键作用,如何特异性地调节其功能而不引起严重不良反应也是需要解决的关键问题。有关神经递质类药物临床研究状况见表1。

表1 治疗阿尔茨海默病(AD)神经递质类药物临床研究[51-52]

续表1
2 tau假说
tau蛋白广泛分布于中枢神经系统的神经元中,参与微管的组装和稳定,对维持神经元的正常形态和保证马达蛋白的胞内运输有重要作用[53]。tau的表达和活性受多种转录后翻译的调控(比如磷酸化[9]、硝化[54]与乙酰化[55]小泛素样因子修饰[54-55])。tau蛋白过磷酸化而导致的异常聚集,最终形成的神经元纤维缠结是AD的一个典型病理特征[58-60]。在AD患者大脑中,tau的磷酸化增加了3~4倍,纤维缠结普遍存在[61],提示tau蛋白聚积对突触失调和神经元丢失的作用。值得注意的是,与可溶性Aβ相似,脑皮质中神经元纤维缠结的密度与AD患者认知障碍的程度正相关[62]。因此,tau也被认为是AD的药物靶点。目前,基于tau假说治疗AD的药物作用机制主要是稳定微管、抑制tau的磷酸化、通过增强自噬作用抑制其异常聚集及使用tau抗体阻止其病理扩散(图1)。
2.1 微管稳定药物
由于tau功能失活导致的微管不稳定是AD的可能病理机制之一,因此,通过补偿tau的功能失活,使微管稳定的一类药物有可能有利于缓解AD患者的症状。紫杉烷类(taxanes)药物TPI 287是紫杉烷双萜类衍生物,与其他大多数紫杉烷类药物不同,它可以通过血脑屏障。此前,另一个紫杉烷类药物epothilone D在具有tau相关病理特征的模型小鼠中,可以通过血脑屏障,并且显著降低前脑tau相关病理以及增加海马脑区神经元的完整性[61-62]。但针对该药物的研究和临床试验在2013年10月终止,试验具体细节没有披露。还有一类可以稳定微管的八肽药物(NAPVSIPQ,NAP)也可以穿透血脑屏障。比如,Allon医疗及Paladin实验室联合开发的davunetide可以与在神经元中特异表达的βIII-微管蛋白相互作用,转基因小鼠吸入davunetide可以有效降低过磷酸化的tau与Aβ多肽水平[65]。然而,davunetide的临床试验目前处于停滞状态,具体原因未被披露。

图1 基于tau假说治疗AD的可能药物靶点[66].HSP90:热休克蛋白90.
2.2 抑制tau的磷酸化
tau含有多个可以被磷酸化的丝氨酸和苏氨酸位点[65-66],这些位点的磷酸化的增强导致tau结合微管的能力下降,同时,更容易组装形成纤维[69]。因此,通过抑制磷酸激酶的活性或增强去磷酸化酶的活性,抑制tau的磷酸化也是一类药物的理论基础。已知有一系列的磷酸激酶参与tau的磷酸化,包括糖原合成酶激酶3(glycogen synthase kinase 3,GSK-3)、细胞周期蛋白依赖性激酶5(cyclindependent kinase 5,CDK5)、丝裂原活化蛋白激酶1(mitogen-activated kinase 1,MAPK1)、蛋白激酶A、p38以及微管亲和调节激酶1-4(microtubule-affinity regulated kinases,MARK1-4)。尽管目前没有确认在AD患者大脑中起主要作用的激酶,但是由于GSK-3和CDK5与神经元纤维缠结显示出高度的共定位,它们的酶活性升高都可以导致tau的过磷酸化,因此被认为是与AD病理相关的tau磷酸化的关键激酶[68-69]。研究发现,这两者之间有相互作用,抑制CDK5活性,会导致GSK-3介导的tau磷酸化增强[72]。这种相互作用进一步增加了tau磷酸化机制的复杂性,因此,若通过抑制这两种酶的活性来抑制tau的磷酸化,必须同时抑制这二者的活性。同时,由于tau某些位点的磷酸化也会阻止纤维形成[73],彻底抑制tau的磷酸化也可能在一定程度上促进纤维的形成。更重要的是,磷酸激酶调控的磷酸化在机体内广泛分布,如何特异性地抑制tau的磷酸化,而不影响磷酸激酶的其他人体正常生理活动所必需的底物仍然是一个巨大的挑战。因此,这一机制仍需要进一步的研究,才能为后续的药物研发提供安全可靠的药物靶点。
由于tau蛋白也受到其他转录后修饰的调节,其中某些调节与磷酸化存在相互作用,因此,抑制磷酸化药物研发的另一个思路是通过调节其他转录后修饰的活性来抑制磷酸化。比如tau的O-GlcNAc糖基化修饰与乙酰化修饰都被证明与磷酸化存在相反作用,抑制正常大鼠O-GlcNAc糖基化酶,可以降低tau的磷酸化[74],而在tau转基因小鼠中,增强tau的乙酰化会抑制tau蛋白的降解,降低tau结合微管的能力也增加其聚集形成纤维缠结的倾向[55-75]。然而,参与调节tau的O-GlcNAc糖基化修饰、乙酰化修饰的酶,都存在多个在其他信号通路中起关键作用的底物,意味着一般的广谱酶抑制剂可能导致多种不良反应,然而针对tau的特异性转录后修饰抑制剂的研发仍然是当前知识水平或技术无法达到的,仍需要进一步的研究。
2.3 抑制tau的聚合及纤维化
由于脑皮质中神经元纤维缠结的密度与AD患者认知障碍的程度正相关[62],所以,抑制tau的聚合及纤维化也是降低其毒性的一种途径。通常认为,tau蛋白的纤维缠结的形成通常是异常折叠的tau蛋白聚集形成晶核,促进正常tau蛋白组装延长形成寡聚体,最终形成纤维缠结[74-75]。因此,通过抑制成核和(或)延长是抑制tau的纤维缠结的两个靶点。目前,唯一具有药物临床应用指标的tau蛋白纤维缠结小分子抑制剂是亚甲蓝(methyleneblue,MB)。体外实验证明,MB可以通过抑制tautau之间的结合,促进tau聚集产物的降解,抑制tau聚集形成神经元纤维缠结,同时,又不影响tau与微管之间的相互作用[76-77]。另外,使用tau转基因小鼠实验证明,MB还具有诱发细胞自我吞噬、神经元保护以及改善认知的作用[80-81]。TRx0237是MB的纯化产物。Ⅰ期临床试验的信息未被披露,而Ⅱ期临床试验在8个月后因为“管理因素”被终止。目前,该药有两项针对AD的Ⅲ期临床试验正在进行中,预计分别将于2015年11月及2016年4月结束。
2.4 增强异常折叠的tau的降解
异常折叠的蛋白通常通过增强泛素-蛋白酶体系与自我吞噬-溶酶体两条通路降解。尽管前者无法水解正常的tau蛋白,但是可以通过热休克蛋白70-热休克蛋白羧基端结合蛋白(heat shock protein-carboxyl-terminus of HSP70 interacting protein,HSP70-CHIP)复合物调节磷酸化的tau的降解[82]。实验表明,在CHIP基因敲除的小鼠体内磷酸化tau的水平上升[83]。而抑制细胞中HSP70-CHIP蛋白复合物调节蛋白HSP90的活性,可以增加HSP70的表达,促进磷酸化tau的降解[84]。转基因小鼠给予可穿过血脑屏障的HSP90抑制剂PU-DZ8进行急性或短期干预,可以降低可溶性和过磷酸化不溶性tau的含量[85]。然而,以HSP90作为药物靶点抑制过磷酸化tau形成纤维缠结的难点在于HSP90在大多数组织中高度表达,它的底物包括调节细胞生长及生存的关键信号通路组分[86]。例如,HSP90敲除对小鼠是致命的,说明通过抑制HSP90的活性促进磷酸化tau的降解很有可能带来严重的不良反应。
除了泛素-蛋白酶体系,自我吞噬-溶酶体通路可以降解过磷酸化形成寡聚体及多聚体的不溶性tau。在过表达人类Tau基因的神经元母瘤细胞中,抑制自我吞噬作用,会降低tau的清除效率[87]。通常使用的增强自我吞噬的药物是雷帕霉素,然而,它会影响包括雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)通路在内的一系列生理活动,包括免疫抑制的作用[88]。尽管对野生型小鼠长期使用雷帕霉素会延长其寿命并降低衰老相关的疾病的发病率[87-88],但是长期影响mTOR通路的活性可导致潜在不良反应,需进一步的研究。此外,tau聚积抑制剂MB诱发的信号通路类似于雷帕霉素[80],具体机制有待进一步研究。另一个自我吞噬增强剂是氯化锂(LiCl),它不仅可以通过抑制肌醇单磷酸酶活性,增强tau的自我吞噬[91],同时也可以通过抑制tau的磷酸化激酶GSK-3的活性,抑制tau的过磷酸化[92],最终降低可溶性和不溶性tau的水平。
2.5 抑制tau的病理扩散
有研究表明,聚集的tau可以在细胞之间传播。异常折叠的tau像“种子”一样,从受损的或将要死亡的神经元释放到细胞间液后,通过细胞的内化作用而进入周围细胞,从而在细胞之间扩散[93]。神经元内无tau纤维缠结的转基因小鼠大脑中注射显示出tau病理的转基因小鼠的大脑分泌物之后,前者出现了tau病理样聚积[94]。若向AD患者注射识别种子tau的疫苗,可否遏制异常折叠的tau在细胞之间的扩散,这一假设促进了以tau为直接靶点的主动与被动免疫疗法的发展。其中,主动免疫是指将载有合成的tau多肽或片段的抗原注入人体诱发宿主的免疫应答反应,产生tau抗体。被动免疫治疗中,tau抗体被直接注入宿主体内,避开了免疫应答过程。主动及被动免疫治疗中,tau抗体可能是通过直接结合tau进行清除或通过标记tau使大脑中的巨噬细胞清除tau。前期动物实验表明,将磷酸化tau多肽作为疫苗注射到P301L的tau转基因模型小鼠体内后,发现小鼠可以产生抗体[95]。在另一tau模型小鼠,THY-Tau22转基因小鼠中的实验结果表明,将含有磷酸化丝氨酸位点422的多肽作为疫苗注入小鼠体内后,接受疫苗注射的小鼠可以产生抗体,同时,其纤维缠结和认知障碍的程度都有所缓解[96]。然而,也有实验表明,使用重组人Tau蛋白作为抗原注射到野生型小鼠体内之后,小鼠出现神经元纤维缠结样结构,并表现出轴突损伤与神经胶质增生[97]。这样截然相反的结果表明,异常折叠的tau通过种子tau扩散到周围细胞,引起tau样病理蔓延这一假说及其机制仍需进一步研究。目前Axon神经科学公司的AADvac-1及杨森的ACI-35都是针对tau蛋白的主动免疫疫苗,它们的Ⅰ期临床试验尚在进行中(表2)。
总之,目前基于tau假说的药物临床试验的情况不容乐观:7个进入临床试验的药物已经有3个被终止了,而只有1个进入了Ⅲ期临床,但由于其安全性问题Ⅱ期临床试验被终止了(表2)。其原因可能是tau在AD病理中的作用被高估了。实际上,在神经元内,可能是Aβ的聚集导致了tau蛋白的过磷酸化而产生了异常聚集,损伤神经突触的正常功能。虽然有证据证明,皮质区tau蛋白纤维缠结的密度而非淀粉样斑块的数目,与神经元的死亡及认知障碍的程度正相关[62],因此有人认为,tau蛋白而非Aβ聚集造成了AD。但是,这一说法忽视了一个重要事实,那就是可溶性Aβ42寡聚体,而非淀粉样斑块,导致了神经毒性和认知障碍[18]。也有研究证明,在人脑脊液中,Aβ的病理改变早于tau相关的病理改变及神经元退行性病变[98]。另外一项研究使用5xFAD的AD模型小鼠,提取其神经元细胞进行三维立体培养,成功还原了脑内淀粉样斑块及tau蛋白导致的神经元纤维缠结。研究结果证明,Aβ可以促进tau的磷酸化。当使用β-或γ-分泌酶抑制剂处理FAD ReN-mAP细胞时,升高的磷酸化tau水平显著下降,尤其是在轴突样结构中,并且磷酸化的tau蛋白阳性的细胞数目也显著降低,这强烈提示磷酸化的tau蛋白在FAD ReN细胞中的聚集是由Aβ聚集引发的[99]。

表2 靶向tau蛋白治疗AD相关药物临床研究[51-52]
3 Aβ假说
淀粉样斑块是AD的一个经典病理特征[100]。1980年代初,由AD患者大脑中的淀粉样蛋白分离出的Aβ多肽证明AD淀粉样老年斑块是由Aβ多肽构成的[7-8]。这也引发了AD的Aβ假说,即Aβ聚集导致的淀粉样斑块是AD的主要致病机制,并引发了其他的病理现象,比如细胞内的神经纤维缠结,突触丢失和神经元死亡。淀粉样斑块的形成由Aβ异常聚集导致,这被认为是Aβ生成与清除之间的动态平衡被打破造成的。因此,基于Aβ假说的药物开发主要围绕降低Aβ生成、防止Aβ聚集和增强Aβ清除三方面进行的。
3.1 抑制Aβ生成
首先,Aβ是β-分泌酶和γ-分泌酶两种天冬氨酸水解酶剪切APP产生的。APP是一种在哺乳动物细胞中广泛分布的约为700个氨基酸的一类跨膜糖蛋白[101]。APP首先被β-分泌酶剪切,分别生成可溶性氨基端sAPPβ以及含有99/89-位点的羧基端片段βCTF。后者随后被γ-分泌酶剪切成AICD和与淀粉样斑块生成相关的可溶性Aβ40和Aβ42[102](图2)。除此以外,APP还可以被α-分泌酶剪切成可溶性的氨基酸sAPP和随后可以被γ-分泌酶剪切成P3和AICD的羧基端,但是这一信号通路通常被认为与淀粉样斑块无关,因此与之相关的研究也较少。总之,Aβ的产生需要β-分泌酶和γ-分泌酶参与剪切,所以目前流行的观点是通过抑制β-分泌酶和γ-分泌酶这两种酶的活性达到抑制Aβ产生的效果。

图2 淀粉样前体蛋白(APP)水解通路相关的AD治疗靶点[102].secretase:分泌酶;Aβ:β淀粉样蛋白;caspase:胱天蛋白酶.
β-分泌酶,通常被称为淀粉蛋白前体蛋白β-分解酶1(β-site APP cleaving enzyme 1,BACE1),是一种天冬氨酸水解酶。尽管BACE1还有一种同系物BACE2,并且两者有51%的氨基酸序列的相似性以及相似的结构[103-105],但是BACE1主要分布在中枢神经系统的神经元中,而BACE2则主要分布在周围组织中[102]。在大脑中,BACE2的表达量明显低于BACE1,且并未被证明是AD相关的β-分泌酶[106-107]。因此,关于Aβ的研究,目前主要集中于BACE1上。自它1999年被发现以来,围绕它与疾病相关的特性进行了深入的研究。本实验室的研究发现,在BACE1的编码区内并不含有任何与AD相关的基因突变[108]。有趣的是,我们还进一步发现,在散发性AD脑中,BACE1的酶活性显著升高[106-107]。这项研究成果为制药工业提供了坚实的理论及实验基础。此外,除了发现BACE1与散发性AD相关,BACE1的功能异常还被发现与精神分裂症和癫痫样行为有关[102-110]。
BACE1是一类约为75 ku的单跨膜蛋白质,在AD大脑中它的蛋白量和酶活性显著升高[106-107]。它的表达和活性受多种转录因子,比如NF-κB[109-110]、过氧化物酶体增殖因子受体-γ(peroxisome proliferator-activated receptor γ,PPARγ)[113]、缺氧诱导因子-1(hypoxia inducible factor-1,HIF-1)[112-114]和转录后翻译,比如糖基化[115-116]、磷酸化[117]、棕榈酰化[118]、泛素化[119]和乙酰化[120]的调控。比如,当BACE1启动子上NF-κB结合位点被激活后,BACE1的表达量会随之增加[111]。另外,各种转录后修饰保证BACE1正常的胞内表达和功能。例如,N型糖基化和乙酰化保证BACE1的成熟和转运[115-120],棕榈酰化可以介导BACE1在细胞膜上的转移并增强其活性[118],而泛素化调控BACE1的降解[119]。任何一种通路的失衡都有可能导致BACE1表达和(或)功能的失调,从而导致Aβ生成异常。
除了APP,BACE1还有多种水解底物,比如神经调节蛋白(neuregulin,NRG)、P-选择素糖蛋白配体(P-selectin glycoprotein ligand 1,PSGL-1)和电压依赖型钠离子通道(voltage-dependent sodium channels,VDSC)等[102]。因此,抑制BACE1的活性,可以抑制APP水解生成Aβ,从而减少Aβ、淀粉样斑块和认知障碍,这可以解释部分抑制BACE1的活性可以降低AD相关脑区的Aβ多肽以及淀粉样斑块含量,并改善损伤的突触可塑性以及学习记忆能力[121]。但是,由于BACE1其他底物的存在,彻底抑制BACE1的活性也会影响其他底物相关的信号通路。比如,最近发表的一项以正常小鼠为对象的研究,证实了低剂量的BACE1抑制剂可以在不影响记忆的情况下阻断Aβ的生成,但是他们发现,高剂量BACE1抑制剂则会导致突触损伤和记忆丢失[122]。另外,BACE1敲除小鼠也显示出空间参考记忆异常以及颞叶相关记忆的损伤[123]。因此,BACE1对学习记忆是有双重效果的,而BACE1抑制剂对AD的疗效与安全性依赖于是否可以适度抑制BACE1的活性。
目前,以抑制BACE1活性为治疗机制的AD药物的临床试验还没有成功的先例。比如,礼来公司于2011年在第十届AD/PD国际会议上宣布:BACEI抑制剂LY2811376由于导致视网膜色素上皮质缺损的可能性而终止于Ⅰ期临床试验。随后,因为可以选择性地抑制BACE1而不影响其他天冬氨酸水解酶,而被寄予厚望的LY2886721,因4例出现肝生化指标异常而终止于Ⅱ期临床试验。然而,这被认为是脱靶效应,而不是因为抑制BACE1本身导致的。
另外,2013年10月罗氏公司也宣布终止BACE1抑制剂RG7129的临床试验而未给出具体原因。但是,目前仍有数个BACE1抑制剂仍处于临床试验阶段并被寄予厚望。尤其是默克公司的MK-8931已经进入Ⅲ期临床试验,前期数据表明,药物基本安全,未见严重不良反应,并且患者脑脊液中的Aβ含量降低高达90%。目前进行的Ⅲ期临床试验不仅将检测该药物对轻度到中度AD患者认知能力的改善,也将检测其对相关生物标志物,如大脑中Aβ的含量、脑脊液中tau蛋白的水平和大脑体积的改变。该临床试验预计于2018年结束。另外,阿斯利康的BACE1抑制剂AZD3293也在7项Ⅰ期临床试验中显示出安全性和显著降低脑脊液中Aβ含量的作用,Ⅱ/Ⅲ期临床试验预计将于2019年结束。除此以外,CoMentis公司的BACE1抑制剂CTS-21166和百健、卫材药业公司的E2609均处于Ⅰ期临床试验中。初期临床试验数据表明药物安全,并且可以降低脑脊液中的Aβ含量,后续试验仍在进行中。但是,BACE1抑制剂BI 1181181正在进行中的Ⅰ期临床试验由于某些受试者出现的皮肤异常而被临时暂停,这可能是由于BI 1181181抑制了BACE1剪切产生黑色素的色素细胞特异性黑色素细胞蛋白,但是由于Vitae未披露试验细节,因此,尚不能确定“皮肤异常”是否与黑色素及PMEL蛋白相关。这些都表明,BACE1或许可以作为一个AD治疗的药物靶点
BACE1抑制剂的临床进展主要受到以下几个方面的限制:①是否可以特异性地抑制BACE1的活性,因为BACE1与其同系物BACE2有51%的氨基酸序列的相似性,如何特异性抑制BACE1的活性而不影响参与其他信号通路的BACE2的功能的特异性抑制剂也是制约BACE1抑制剂开发的重要因素之一。②由于BACE1在大脑中过度切割APP,因此,BACE1抑制剂必须通过血脑屏障进入大脑抑制BACE1的效果。但是血脑屏障允许通过的最大分子质量约为500 u[124]。因此,开发小分子BACE1抑制剂将是疗效的关键之一。BACE1抑制剂P10-P4、OM99-2以及OM00-3均因为分子过大无法通过血脑屏障,或半衰期太短等因素导致无法进入临床试验[125]。此外,值得关注的是,BACE1酶活性区较大,大多数抑制BACE1的小分子不能有效的抑制BACE1酶活性。因此,深入研究BACE1在AD发病机制中的作用及相关信号通路的调控将是研发安全有效的抑制BACE1酶活性的小分子化合物的必要条件。
γ-分泌酶是由衰老蛋白1(presenilin1,PS1)和PS2、呆蛋白(nicastrin)、Aph-1和PS增强子2一起构成的复合物[126-127]。PS的基因缺失会显著性降低γ-分泌酶的活性,暗示它是γ-分泌酶的重要活性位点[128]。PS1和PS2的错义基因突变会特异性地改变γ-分泌酶对APP的剪切,导致Aβ42含量的升高从而加速淀粉样斑块的形成,最终导致家族性的AD[129]。因此,另一类治疗AD药物作用机制在于抑制γ-分泌酶,主要是早老素蛋白的活性,从而降低Aβ生成,防止或减缓淀粉样斑块的形成。但是,γ-分泌酶的底物并不限于APP,还包括在其他关键信号通路中对发育有重要作用的蛋白,比如Notch、ErbB4、E-钙黏着蛋白、肝配蛋白-B2和CD44[130-131],所以抑制γ-分泌酶的药物会影响其他信号通路的正常生理功能,尤其是当这种影响无法通过其他通路得到补偿的时候。比如Notch是调控细胞增殖和分化关键信号通路的重要组分,其正常发育依赖于γ-分泌酶对它的剪切[132]。因此,抑制γ-分泌酶的药物会降低Notch的活性,导致其产生多种严重不良反应甚至患者死亡。其中,礼来公司的γ-分泌酶抑制剂Semagacestat由于导致患者罹患皮肤癌的概率上升、感染以及疗效不足,甚至给药组患者的认知功能发生恶化等原因,于2012年终止临床试验[133]。而未产生显著不良反应的药物也未能有效缓解AD症状,比如氟比洛芬(R-flurbiprofen)由于未达到改善患者认知和日常生活能力的预期效果而终止于Ⅲ期临床试验。在γ-分泌酶抑制剂均以失败告终之后,当前以γ-分泌酶为靶点的AD治疗药物主要集中在调整其功能,而非简单地抑制其酶活性。比如当前处于Ⅱ期临床试验的γ-分泌酶调节剂EVP-0962的作用机制是在不影响γ-分泌酶对Notch正常剪切的情况下,通过改变它的剪切位点,使其水解产生Aβ38或其他更短的多肽,而非与AD相关的Aβ40,或Aβ42,从而达到抑制淀粉样蛋白聚集的效果。同样处于Ⅱ期临床试验的NIC5-15在不影响对Notch正常剪切的情况下,通过调节γ-分泌酶的功能降低Aβ的生成。但是,由于基于选择性抑制γ-分泌酶产生Aβ42的两种药物的Ⅱ期临床试验尚未完成,因此,基于这一机制抑制Aβ聚集和淀粉样斑块形成的可行性仍有待证明。
3.2 抑制Aβ聚集
除了通过抑制Aβ的生成,还可以通过抑制Aβ的聚集而减少淀粉样斑块的形成。由于在老化大脑中,有生物活性的金属离子浓度的升高会促进淀粉样斑块的形成以及具有神经毒性的氧化反应[132-133]。因此,一部分以抑制Aβ的聚集为作用机制的药物靶点是金属离子。比如,PBT2是一种金属-蛋白衰减复合物,通过将铜锌离子转运到细胞内,从而减少胞外离子浓度,干扰金属与Aβ多肽之间的相互作用而防止Aβ聚集。尽管它被证明可以有效改善APP转基因小鼠的突触树突棘密度和相关蛋白含量[134-136],但是在一项Ⅱ期临床试验中,它未能显示比安慰剂更显著地降低Aβ聚集的效果,但进一步的数据仍有待分析和公布。另外,除了Aβ的生成增加,与对照组相比,晚发型AD患者的中枢神经系统中Aβ42和Aβ40的清除速率都显著降低[137]。这也导致了Aβ产生与清除的动态平衡被破坏,从而产生淀粉样沉淀。因此,促进Aβ清除也是抑制淀粉样斑块形成的另一手段。
3.3 促进Aβ清除
体外证据表明,Aβ可以被多种水解酶降解,其中最受关注的两种水解酶是胰岛素降解酶(insulin-degrading enzyme,IDE)和脑啡肽酶。IDE是由神经元分泌的锌金属蛋白酶,分布在细胞质、细胞器膜和细胞表面,因此可以降解细胞膜内和膜外的可溶性的Aβ单体,剪切产物无神经毒性并且不易聚集形成斑块[136-137]。脑啡肽酶是轴突与突触膜锚定蛋白,它的催化部位朝向膜外。因此,主要剪切膜外的Aβ42,并且,它既可以剪切单体Aβ,也可以剪切Aβ寡聚体[139]。
此外,Aβ自身也是AD治疗的一个主要药物靶点。以Aβ为直接靶点的主动与被动免疫疗法,主要针对产生神经毒性的Aβ42。在既往的实验中,免疫治疗在小鼠及非人类灵长动物中都取得了较好的效果。比如,在1999年,Schenk等[140]首先发现对AD模型PDAPP转基因小鼠在AD病理症状出现之前进行Aβ42免疫,可以防止其出现淀粉样斑块,而在AD晚期进行免疫,也可以显著降低AD样病理的程度及进展。随后,在PDAPP小鼠中进行的被动免疫实验也表明,外周注射Aβ抗体可以通过血脑屏障、减少Aβ聚积、加速Aβ降解[141]。此后,在多种AD模型小鼠中进行的实验都表明,Aβ主动及被动免疫治疗可以明显减少Aβ聚集,改善认知障碍[142-146]。由于恒河猴、绿猴的大脑有与人类相似的年龄相关的Aβ聚集,因此,成为研究人AD发病机制及药物作用的良好模型。在恒河猴和绿猴中进行的2项实验也都表明,Aβ主动免疫疫苗可以显著增加血浆中Aβ含量[147-148]。绿猴接种疫苗后,脑脊液中Aβx-40多肽含量显著减少至64%,大脑中不溶性Aβx-42多肽含量显著减少66%,额皮质区也出现了具有Aβ42免疫活性的斑块[148]。这些都表明,Aβ作为免疫靶点在AD治疗上的前景。2000年,由伊兰和惠氏公司主导的Aβ疫苗AN-1792及佐剂QS-21在英国进行了单剂量和多剂量的Ⅰ期临床试验。结果表明,有59%接受多剂量注射的受试者出现抗Aβ反应,并且该药耐受性良好,无不良反应。然而,在后续涉及到300例中重度AD患者及72例健康受试者的Ⅱa期临床试验中,约有6%(18例)的患者在接种Aβ疫苗之后出现了脑膜炎[147-148]。研究发现,这一不良反应与抗体反应无关,主要是与细胞毒性T细胞的激活相关及自身免疫反应相关[151-154]。更准确地说,辅助型T细胞1(T lymphocyte helper-1,Th1)是造成该不良反应的主要原因[154]。在最大限度内降低Th1的不良免疫反应是主动免疫型Aβ疫苗必须要解决的问题。首先,在人类Aβ片段中,T细胞主要的免疫氨基酸位点是16-33[155]。已经有实验证明,通过使用较短的氨基端Aβ片段作为疫苗可以有效减少AD模型APP转基因小鼠的Th1型不良反应[156-158]。比如,目前处于Ⅱ期临床试验中的AFFiRiS AG公司的Affitope AD02就是模拟Aβ氨基端前6个氨基酸的合成多肽。它不含主要的T细胞相关的氨基酸位点,因此可以产生Aβ抗体而不引起Th1类免疫反应。它的Ⅰ期、Ⅰb期以及Ⅱ期临床试验表明,试验基本安全、耐受性良好、无脑膜炎病例出现,但是未达到主要和次要的疗效判定指标。进一步的试验仍在招募受试者。也有研究表明,在AN-1792的临床试验中使用的佐剂QS-21会造成Th1型不良反应[151],因此,通过采用其他佐剂避开Th1相关的不良反应也是Aβ主动疫苗的研究方向之一。另外,通过改变给药方式也是一种加强Aβ疫苗疗效的方法。比如,使用鼻腔给药代替静脉注射Aβ疫苗可以增强表位特异性的Aβ抗体产生的效率[159]。此外,采用被动免疫方法直接注射Aβ抗体避开T细胞相关的自身免疫反应也是目前Aβ疫苗的研究方向。目前处于临床试验中的14个Aβ疫苗中有11个都属于被动免疫疗法。比如,中外制药、霍夫曼罗氏集团的gantenerumab以及礼来公司的solanezumab都已经处于Ⅲ期临床试验。gantenerumab是人类免疫γ球蛋白1(immune γ globulin 1,IgG1)抗体,可以结合Aβ淀粉样斑块。体外实验表明,gantenerumab可以激活人类小胶质细胞的吞噬作用清除AD患者脑内的Aβ,还可以中和Aβ42寡聚体造成的大鼠的LTP损伤[160]。目前已经结束的Ⅰ期临床试验结果证明,其基本安全且耐受性良好。然而,在200 mg的高剂量组的6例患者中,有2例病灶区出现了短暂的炎症反应和血管性水肿,说明Aβ相关的影像学异常在后续的试验中值得注意。2014年12月,罗氏宣布基于期间无效分析数据,gantenerumab的一项Ⅱ/Ⅲ期临床试验被终止。因为未有不良反应的进一步报道,这项试验的终止很有可能是因为gantenerumab疗效不足或脱靶效应。目前,尚有另一项针对中度AD患者的gantenerumab的Ⅲ期临床试验仍在进行中。然而鉴于其之前由于无效分析而终止的试验,此项Ⅲ期临床试验的前景也不容乐观。由于gantenerumab主要是针对已形成的淀粉样斑块,此时,即使gantenerumab可以减少斑块数量,可是已经无法逆转Aβ的神经毒性已经造成神经元损伤。但是,礼来公司开发的solanezumab也是人类IgG1抗体,它可以识别可溶性Aβ。Ⅰ和Ⅱ期临床试验证明其基本安全,耐受性良好,未出现炎症反应、微出血和血管性水肿等严重不良反应。虽然,Ⅱ期临床试验表明,solanezumab对血浆及脑脊液中的Aβ水平的增强效果是剂量依赖的。但是,在2012年,2项Ⅲ期临床试验结果表明,与安慰剂相比,solanezumab对认知功能没有改善,然而它减少了轻度AD患者的认知能力的损伤。针对这一结果,2013年7月,礼来公司开始了针对轻度AD患者的Ⅲ期临床试验,该试验将于2016年12月结束。目前,显性遗传AD互助组织(Dominantly Inherited Alzheimer Network,DIAN)开展了为期5年、联合应用gantenerumab与solanezumab预防常染色体显性突变的遗传性AD的Ⅱ/Ⅲ期临床试验。因此,目前虽然没有强有力的证据表明Aβ疫苗可以治疗AD,但是临床试验结果表明,在出现明显病理特征之后接种Aβ疫苗可能无效,而在出现轻微症状甚至之前,接种疫苗可以在一定程度上改善认知。这可能是由于在AD早期,可溶性Aβ已经开始造成神经毒性,而当出现典型病理特征,如淀粉样斑块及神经元死亡之后,再接种疫苗,已经无法逆转这种情况了,因此,gantenerumab与solanezumab的临床试验表明,它们无法改善中度AD患者的认知。以Aβ为靶点治疗Aβ相关临床药物研究见表3。

续表3
4 炎症假说
如前所述,目前以Aβ和tau这两种蛋白为直接靶点的AD治疗药物尚无一种通过临床试验。事实上,AD的发病机制非常复杂,除了Aβ和tau蛋白直接导致的标志性病理变化外,尚存在其他重要机制。炎症反应是机体免疫系统对有害刺激如病原体、死亡细胞、创伤等产生的保护性生理应答,涉及非常广泛的细胞、分子机制,形成复杂的调控网络。神经炎症反应在清除脑内残骸、应答病原体入侵等发挥重要作用,但如果炎症反应失控或长期存在,将会对大脑产生损害,这种情况在神经退行性疾病中同样存在[159-160]。包括本实验室在内的大量研究工作显示,AD脑中胶质细胞被激活,大量的免疫补体蛋白和免疫细胞因子等被释放[163-168]。因此AD发病机制的炎症假说也日益受到关注[167-168]。
4.1 不同细胞的参与
4.1.1 小胶质细胞
小胶质细胞占大脑细胞数目的10%~15%[171],能清除脑内死亡的神经元和入侵的感染源等[172]。AD患者和AD模型小鼠中均发现小胶质细胞激活且聚集于Aβ斑块周围[173],而关于小胶质细胞激活是促进或抑制AD发生发展尚存在争议,这两种相反的作用取决于疾病所处的状态和受影响的脑区[174]。
一方面,激活的小胶质细胞聚集于Aβ斑块周围就已经表明小胶质细胞尝试通过噬菌作用清除Aβ斑块[175],尽管实验证实小胶质细胞并不能有效降解其内吞的Aβ[176-178]。而且小胶质细胞可能通过释放胰岛素降解酶清除Aβ[179-181]。髓样细胞表达的2型触发受体(triggering receptor expressed on myeloid cells 2,TREM2)是一种表达于小胶质细胞等髓样细胞表面的受体蛋白[182],最近2篇报道关于TREM2敲除对AD模型小鼠症状的影响虽截然相反,但均发现Aβ斑块周围激活的小胶质细胞数目减少[183-184]。Wang等[183]的报道进一步说明TREM2的R47H位点突变能减弱TREM2识别与Aβ相互作用的带负电的脂质体的能力,进而损伤小胶质细胞对Aβ聚集的响应,破坏其抑制神经退化的能力,这也为TREM2的R47H突变使AD患病风险增加[185-187]提供了可能的解释。最近一项非常有趣的研究报道了超声能清除AD转基因模型APP23小鼠脑内Aβ斑块并恢复小鼠的记忆能力,免疫组化分析显示,小胶质细胞被激活且内吞Aβ增加[188]。以上结果均显示,小胶质细胞激活在逆转AD病理病症中发挥积极的作用。
另一方面,年老的AD转基因模型APP/PS1小鼠脑内小胶质细胞清除Aβ的能力下降[189];Krabbe等[190]报道了AD模型小鼠中小胶质细胞的噬菌活性等功能被严重损害的程度与Aβ斑块正相关,利用Aβ免疫减少斑块,能恢复小胶质细胞的噬菌能力。小范围(共37名被试,其中13人为AD患者)的正电子放射断层成像监测显示,小胶质细胞激活程度与认知能力呈负相关[191]。而CX3CR1,一种小胶质细胞特异性表达的、介导神经元-小胶质细胞通讯的趋化因子受体被敲除后,能阻止AD模型小鼠的神经元丢失[192],此外,Aβ聚积及其周围被激活的小胶质细胞数目均有所减少[193],这可能是由于CX3CR1敲除改变了神经元与小胶质细胞之间的通讯,从而影响了小胶质细胞的激活与吞噬能力。
目前正在进行的以小胶质细胞为靶点的临床试验主要有3项:CHF 5074(临床Ⅱ期)、GC 021109(临床Ⅰ期)和沙格司亭(临床Ⅱ期),其共同机制均为激活小胶质细胞,增强其噬菌能力,进而清除Aβ斑块,虽然对于CHF 5074而言,尚存在其他可能作用机制,如恢复神经发生、调节突触可塑性[194]等。
4.1.2 星形胶质细胞
星形胶质细胞是脑内重要的神经支持细胞,能分泌回收神经递质[195]、维持胞外介质离子平衡[196]、调节能量代谢等[195-196]。同小胶质细胞一样,在AD患者脑内[162]和AD模型小鼠脑内[199]均发现星形胶质细胞激活,其作用也呈现出两面性。一方面,激活的星形胶质细胞有利于清除Aβ和保护神经元[198-199]。AD转基因模型APP/PS1小鼠敲除星形胶质细胞激活所必需的两种蛋白——胶质细胞原纤维酸性蛋白(glial fibrillary acid protein,GFAP)和波形纤维蛋白(vimentin,Vim),导致Aβ斑块增加[202]。最新的研究报道表明,星形胶质细胞和神经元共培养能保护神经元免受Aβ毒性损害[203]。而另一方面,激活的星形胶质细胞也被证实具有神经毒性作用。如激活的星形胶质细胞能通过释放抑制性神经递质GABA,进而损伤AD模型鼠的学习记忆能力[204]。而在尚未出现严重Aβ病理的7到8月龄APP/PS1小鼠海马中双侧注射抑制星形胶质细胞细胞激活的慢病毒,发现Aβ减少,突触可塑性和认知能力均增强[205]。
4.1.3 神经元
以往观点认为神经元不参与神经炎症反应,但越来越多的证据表明,神经元自身能分泌一些炎症调节因子,也就是“神经元免疫或补体系统”(neuronal immune or complement system),如免疫补体系统的全套因子[206]。细胞实验证实,Aβ能激活神经元补体系统,产生攻膜复合体(membrane attack complex,MAC)攻击细胞,如果神经元缺乏内源性补体调节因子CD59,将加剧神经元死亡[207]。而AD患者脑内发现补体因子mRNA较正常对照组明显增高[208],CD59蛋白水平明显降低[165]。其他一些炎症因子如环氧合酶和巨噬细胞集落刺激因子等在AD患者神经元中的表达均有升高[207-208]。
此外,有人提出“神经元致神经炎症”(neurogenic neuroinflammation)概念,以此来描述机体响应过度的神经元活动所产生的免疫应答[211],但尚不清楚这种由神经元活动导致的炎症反应在AD中的作用。
4.2 分子调节因子
有许多细胞因子参与了神经炎症反应,以往将炎症相关因子简单分为抗/促炎因子,但正如Wyss-Coray[212]指出的,抗/促炎的概念仅适用于描述特定时间细胞或蛋白的作用,因为一些细胞因子除了参与炎症反应外尚有其他功能,如肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)除了能促进炎症反应,还能调节突触递质传递[213]。再如TGF-β1在免疫应答前后对炎症反应的作用是相反的[214]。
TNFα是一种与全身免疫相关的细胞因子,具有广谱生物活性,在许多免疫相关疾病中起关键作用,且被广泛认为是一类抗炎和免疫抑制药物分子作用的靶点,其抑制剂对自身免疫性疾病的药理作用已广为人知[213-214]。我们发现,TNFα及其Ⅰ型受体(TNFRⅠ)在AD脑中表达较正常对照组显著升高,且TNFRⅠ表达水平升高伴随神经炎症的出现发生在神经元死亡之前数年,而TNFRⅡ表达却显著降低[217]。
TNFα的2种受体在脑内细胞中差异性表达,其中TNFRⅠ在神经元表达较多,而TNFRⅡ在胶质细胞尤其是星形胶质细胞表达较丰富,两者功能也截然相反,TNFRⅠ含一个能激NFκB信号通路并导致细胞凋亡的胞内区域——死亡结构域(death domain,DD)[218],一旦TNFRⅠ与其配体结合,DD就会启动凋亡相关信号通路引起细胞死亡。在AD中,脑内激活的胶质细胞分泌TNFα,活化TNFRⅠ,导致神经元死亡[219]。而TNFRⅡ的分子不含DD[220],TNFRⅡ基因敲除导致神经元更容易死亡和提早出现AD样病理[166-221],表明TNFRⅡ在神经元存活中起营养或保护作用,尽管其具体作用机制尚不清楚。
我们猜测神经系统中的2种TNFR通过脑内不同的细胞以及不同的信号传导通路保持脑内神经-免疫调节的平衡,而这种平衡被打破可能是AD脑内出现免疫炎症和突触变性,直至神经网络功能异常的原因之一。因此,阐明TNFR介导的AD脑内分子、细胞间调控机制将有助于寻找新的AD治疗靶点[220]。
Birch等[222]总结了关于调控炎症因子对AD模型小鼠的AD病理症状的影响。表4列出了最近的、在Birch综述中未述及的几项关于炎症因子的研究。
4.3 补体系统
补体系统是先天性免疫组成部分,在机体免疫应答中起重要作用,如清除入侵病原、凋亡细胞和组织细胞残骸等[231-232]。如图3所示,补体系统的激活有3条途径:经典途径、凝集素途径和替代途径,且以C3为中心,最终产生C5b-9复合体,或称MAC。MAC对细胞膜有致孔作用,能导致细胞裂解死亡[234]。但是,补体系统异常激活能加剧炎症反应,产生细胞死亡等破坏性作用,且与多种人类疾病相关[235]。关于补体系统在AD中的作用,近来的综述文献已有较好的总结[165,204,235-236]。补体系统在神经退行性疾病中有双重作用,它可以清除神经系统中的有害物质,比如Aβ与tau蛋白,从而促进细胞再生;也会产生炎症导致组织损伤。其正常功能的发挥依赖于各组分之间的相互作用的平衡,这与中华文化的“阴阳互补”观念很相似,“complement”正是“互补”之意(图4)。而这种平衡被打破在AD发病发展中可能起重要作用[206]。
近来关于AD中补体系统的研究取得一些新的进展。如Lian等[168]报道了NF-κB通过激活星形胶质细胞释放C3,作用于神经元上的C3a受体,损伤神经元形态和功能;而抑制C3a受体可改善AD模型小鼠的认知障碍,暗示神经元上的C3a受体可能成为AD治疗的新靶点。
4.4 临床研究
尚无基于炎症假说的AD临床试验成功实例,正在进行的有近10项(表5),而宣告失败的已有数十项,其中有5项是非甾体类抗炎药(nonsteroidalantiinflammatory drugs,NSAID),分别是氟比洛芬(flurbiprofen)、布洛芬(ibuprofen)、罗非昔布(rofecoxib)、萘普生(naproxen)和塞来昔布(celecoxid)。NSAID被认为可抑制环加氧酶2(cyclo-oxygenase-2,COX-2)的活动性,减少前列腺素的合成,进而减轻炎症反应。
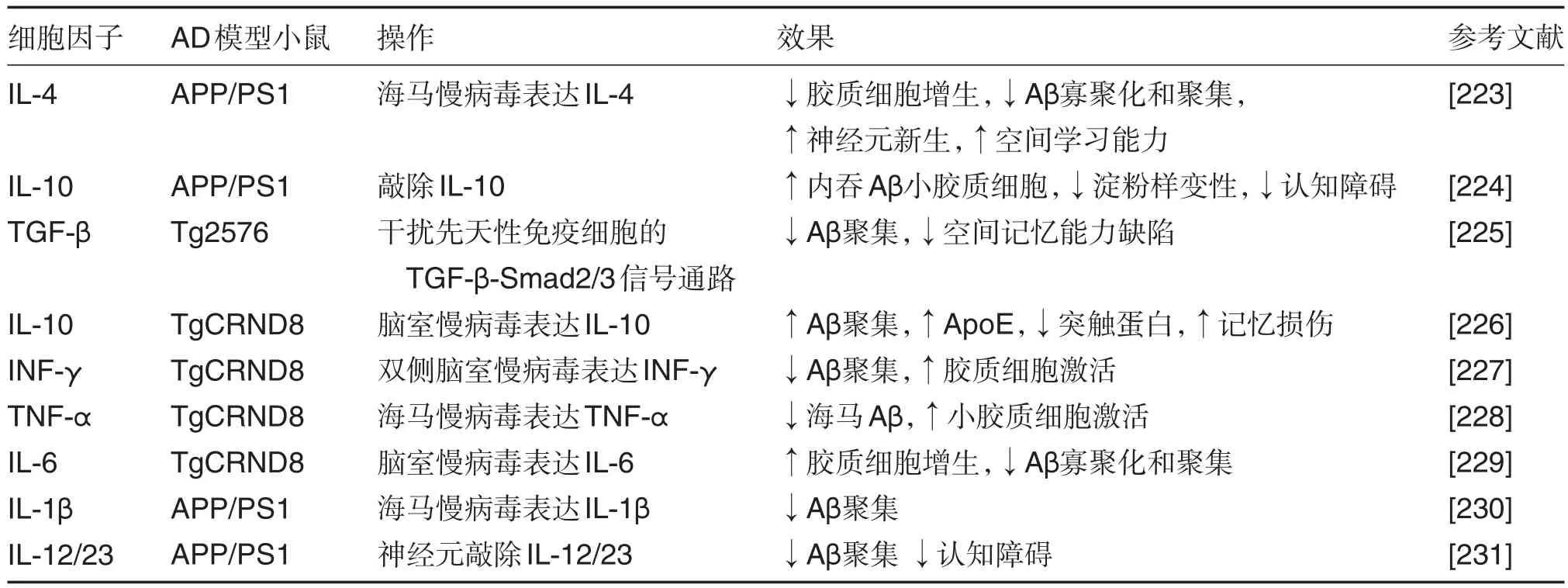
表4 与AD发病机制相关的炎症因子
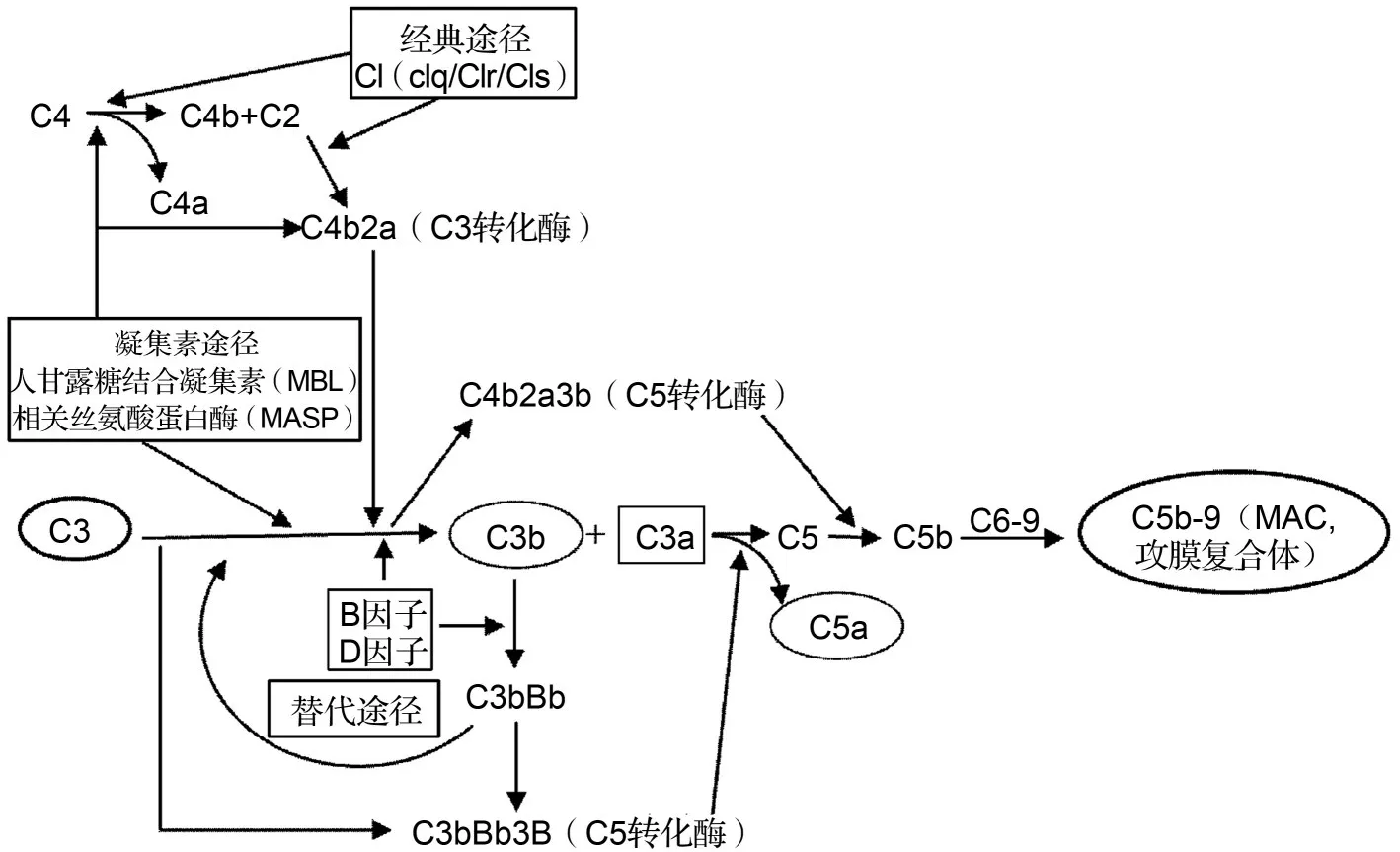
图3 补体系统激活通路[235].

图4 补体系统在AD中的阴阳作用[206].
流行病学的研究结果显示,长期服用NSAID治疗炎症能降低AD患者风险或保护认知[238-240],研究人员开始对用NSAID治疗AD产生兴趣,也因此有了不同NSAID如布洛芬、萘普生和罗非昔布等用于AD的临床试验研究。AD模型细胞和动物实验也证实,某些NSAID能减少Aβ[240-241]。
罗非昔布是美国默克公司制造、出售的一种NSAID,由美国FDA于1999年5月20日批准上市。默克公司对罗非昔布开展了3次AD临床试验,均以失败告终。在2000年和2001年,第1项在351名轻度至重度AD患者中开展的临床试验发现,与安慰剂组相比,罗非昔布给药组未能延缓AD患者的认知衰退。在2004年,默克科研人员公布了第2项临床试验报告,此次试验有692名为50岁以上的轻度至中度AD的患者参加,最终有70%完成了为期12个月的试验,而且罗非昔布对认知衰退等临床症状均无任何改善。同年9月,默克公司宣布,考虑到罗非昔布可能导致严重的心血管损伤进而引发心脏病或卒中,决定从市场上撤回罗非昔布。第3项旨在检测罗非昔布能否延缓AD发病的临床试验选取了725名轻度认知障碍作为受试者,结果也失败了。
另外2个失败的NSAID——萘普生和塞来昔布均进行了2次临床试验(与塞来昔布不同的是,萘普生能抑制COX-1和COX-2,但它们的抗炎机制与其他NSAID一样,都是抑制前列腺素的合成),其中一次是同时进行的,即AD抗炎预防试验(Alzheimer Disease Anti-Inflammatory Prevention Trial,ADAPT),旨在确定这两种药能否延缓AD发病或老年相关的认知衰退。此项临床试验由美国国立老年研究所(National Institute on Aging,NIA)资助,招募了2625名70岁以上、父母或兄弟姐妹患有AD或其他老年性痴呆的被试者,于2001年开始,拟进行5~7年。试验仅开展1年即受到公众关于这两种药物的临床试验依据不足的质疑并被建议暂停。试验进行到2004年,默克公司宣布将另一类NSAID药物——罗非昔布撤出市场;NIA随即终止了萘普生和塞来昔布的ADAPT试验,称萘普生增加了心血管不良反应。而基于有效的试验数据分析,萘普生和塞来昔布均未能延缓痴呆发生和认知衰退,其中萘普生甚至能轻微地加速认知衰退。而2000年和2001年进行的关于萘普生治疗AD的临床试验未发现其有助于延缓认知衰退。但是,2000和2005年,在美国加利福尼亚大学洛杉矶分校进行的临床试验表明,塞来昔布也许能改善伴有老年相关记忆衰退人群的认知能力、增加局部脑代谢。但是此次试验只有88名受试者,均值年龄为58.7岁,且仅仅是自报有轻度记忆障碍但记忆能力测试分数正常[243]。

表5 靶向炎症因子治疗AD相关药物临床研究[51-52]
虽然基于炎症假说的AD治疗药物已有诸多失败案例,但现在仍在进行中的临床试验也带来一线希望,如CereSpir™和Chiesi Pharmaceuticals共同开发的CH5074在Ⅱ期临床试验中显示,其能减少受试者脑脊液中炎症因子CD40和TNF-α的浓度,且能改善携带ApoE4基因受试者的行为能力。而健赞和赛诺菲开发的沙格司亭,是一种合成的粒细胞-巨噬细胞集落刺激因子(granulocyte-macrophage colony-stimulating factor,GM-CSF)糖蛋白,已被美国FDA批准用于骨髓移植后新生单核粒细胞和巨噬细胞等,而用于AD临床试验的依据是其可能增加小胶质细胞等吞噬Aβ的能力并激活其他神经保护性的先天性免疫应答。GM-CSF能减轻AD模型小鼠的淀粉样病理改变,增加小胶质细胞数目,改善认知[244]。2013年,NIA奖励支持了沙格司亭的一项Ⅱ期临床试验。另外,本实验室发现,对AD模型APP23小鼠长期使用小分子抗炎药沙利度胺可以显著降低其大脑中的BACE1酶活性,减轻Aβ病理改变[245]。目前,由Celgene公司主导的沙利度胺的临床试验处于Ⅱ/Ⅲ期。此项为期24周的临床试验将涉及到20例受试者,旨在研究沙利度胺是否可以显著降低轻度及中度AD患者血浆及脑脊液中AD相关的生物标志物的含量。
5 结语
迄今为止,虽然已经有大量的AD药物临床试验以失败告终,同时已经被批准用于临床的药物的疗效难以令人满意,但是,AD研究专业论坛Alzforum数据库中的资料显示,目前尚处于临床试验中的AD药物有82个,其中处于Ⅲ期及Ⅳ期临床试验的药物达18种。比如,默克公司的MK-8931,它作为BACE1抑制剂已经进入Ⅲ期临床试验,前期试验被证明可以安全有效地减少患者脑脊液中高达90%的Aβ。这些都为AD的临床治疗留下了希望。
目前AD药物研发领域的困境主要是由于以下3个原因:①药物缺乏临床应用指标,比如难以通过血脑屏障,不良反应,易产生抗性等;②由于AD复杂的病理机制和临床表现,目前没有合适的动物研究模型;③现有假说,很多关键机制都未被完全揭示。
尽管AD的发病机制被大致分为4大假说,但是实际上,它们之间存在多种形式的相互作用,这进一步加大了AD的复杂性。比如,星形胶质细胞分泌GABA递质损伤AD模型小鼠的学习记忆能力[204],这表明了神经系统与免疫系统的相互作用。值得注意的是,本实验室发现,可以改善Aβ病理的小分子抗炎药沙利度胺是通过抑制BACE1活性减少Aβ生成[245],这表明在Aβ假说中,免疫系统除了参与Aβ的清除,也调控Aβ的生成。又比如,抑制GSK-3调节的磷酸化可以抑制γ-分泌酶调控的Aβ生成,同时抑制tau的过度磷酸化[246],这说明Aβ与tau的磷酸化之间存在相互影响,抑制GSK-3可能可以同时缓解淀粉样斑块和神经元纤维缠结。总之,AD的病因可能不是单一机制造成的。因此,寻找安全有效的AD治疗方案可能需要综合神经元保护假说、tau假说、Aβ假说以及炎症假说。另外,目前用于AD研究的动物模型大多是针对单一假说构建的转基因小鼠,它们的病理成分较为单一。根据Alzforum数据库中的资料,当前主要的AD转基因小鼠模型有113个,其中只有24个有多个基因的改变,并且这24种模型小鼠中的大多数是通过改变编码APP及γ-分泌酶的基因构建的。因此,它们都只能较好地反映Aβ假说相关的病理改变。所以,即使不考虑小鼠与人类之间的物种差异,当前已建立的AD动物模型也不够理想。好的动物模型的建立将加速AD病理机制的研究,是解答以下关键问题的基础。比如,Aβ多肽多聚体及tau蛋白是如何被错误折叠而产生异常聚集的,它们是通过怎样的信号通路在大脑中扩散并影响认知的,在这一过程中,免疫系统和神经网络又起着什么样的作用。以及除了经典的四大假说之外,其他因素对AD发病和病程的影响,比如目前为止发现的晚发型AD的最大基因风险因子APOE4[247]、生物能应激、脑外伤和生活习惯等,都在AD的发病过程中起作用。虽然老年或是衰老是AD的最大危险因素,但这种“老化”、“衰老”开始的时间究竟是生物学上的“老年”或者“中年”,还是实际上“青年”、“少年”就已经开始发生了。这将是一个不可忽视的重要课题。
随着科技发展,以上限制AD机制研究及药物研发的桎梏都有可能被打破。比如,高通量筛选大大提高了寻找新型药物的效率;CRISPR/Cas等新型基因编辑技术的兴起大大加速了转基因小鼠的构建效率,这将有利于建立能较好反映AD患者病理情况的AD模型小鼠的构建。同时,越来越多新的技术手段被用于AD早期诊断与后期干预。比如,虽然到目前为止,并没有针对脑部病变的生物标志物可以用来准确地进行AD早期诊断[51],但是随着在体脑部成像技术的发展,结合生物标志物有可能早期诊断AD。另外,利用扫描超声短暂打开血脑屏障可以显著降低AD模型小鼠大脑中Aβ的含量[188]。这些为AD的临床治疗带来了新的希望。
致谢:感谢中国科学技术大学生命科学学院薛天教授、刘强教授和张智教授给予的鼓励和建议。
[1]Prince M,Albanese E,Guerchet M,Prina M. The global observatory for ageing and dementia care[EB/OL].[2015-03-01].http://www.alz.co. uk/research/WorldAlzheimerReport2014.pdf
[2]Wimo A,Prince M.The global economic impact of dementia[EB/OL].[2015-03-01].http://www. alz.co.uk/research/files/WorldAlzheimerReport 2010.pdf
[3]Barnes DE,Yaffe K.The projected effect of risk factor reduction on Alzheimer's disease prevalence[J].Lancet Neurol,2011,10(9):819-828.
[4]Rosendorff C,Beeri MS,Silverman JM.Cardiovascular risk factors for Alzheimer's disease[J].Am J Geriatr Cardiol,2007,16(3):143-149.
[5]Sharp ES,Gatz M.Relationship between education and dementia:an updated systematic review[J].Alzheimer Dis Assoc Disord,2011,25(4): 289-304.
[6]Van Den Heuvel C,Thornton E,Vink R.Traumatic brain injury and Alzheimer's disease:a review[J].Prog Brain Res,2007,161:303-316.
[7]Glenner GG,Wong CW.Alzheimer's disease and Down's syndrome:sharing of a unique cerebrovascular amyloid fibril protein[J].Biochem Biophys Res Commun,1984,122(3): 1131-1135.
[8]Masters CL,Simms G,Weinman NA,Multhaup G, McDonald BL,Beyreuther K.Amyloid plaque core protein in Alzheimer disease and Down syn-drome[J].Proc Natl Acad Sci USA,1985,82(12):4245-4259.
[9]Lee VM,Goedert M,Trojanowski JQ.Neurodegenerative tauopathies[J].Annu Rev Neurosci, 2001,24:1121-1159.
[10]Latta CH,Brothers HM,Wilcock DM.Neuroinflammation in Alzheimer's disease;A source of heterogeneity and target for personalized therapy[J].Neuroscience,2014,5,S0306-4522.
[11]Smith MA,Perry G,Richey PL,Sayre LM, Anderson VE,Beal MF,et al.Oxidative damage in Alzheimer's[J].Nature,1996,382(6587): 120-121.
[12]Mucke L,Selkoe DJ.Neurotoxicity of amyloid βprotein:synaptic and network dysfunction[J].Cold Spring Harb Perspect Med,2012,2(7): a006338.
[13]Gómez-Isla T,Price JL,McKeel DW Jr,Morris JC, Growdon JH,Hyman BT.Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease[J].J Neurosci,1996,16(14):4491-4500.
[14]Sisodia SS.Alzheimer's disease:perspectives for the new millennium[J].J Clin Invest,1999, 104(9):1169-1170.
[15]Tosun D,Joshi S,Weiner MW.Neuroimaging predictors of brain amyloidosis in mild cognitive impairment[J].Ann Neurol,2013,74:188-198.
[16]Selkoe DJ.Alzheimer's disease is a synaptic failure[J].Science,2002,298(5594):789-791.
[17]Spires-Jones T,Knafo S.Spines,plasticity,and cognition in Alzheimer's model mice[J].Neural Plast,2012,2012:319836.
[18]Walsh DM,Klyubin I,Fadeeva JV,Cullen WK, Anwyl R,Wolfe MS,et al.Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiationin vivo[J].Nature,2002,416(6880):535-539.
[19]Levey AI,Kitt CA,Simonds WF,Price DL, Brann MR.Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies[J].J Neurosci, 1991,11(10):3218-3226.
[20]Gu Z,Zhong P,Yan Z.Activation of muscarinic receptors inhibits beta-amyloid peptide-induced signaling in cortical slices[J].J Biol Chem, 2003,278(19):17546-17556.
[21]FisherA,PittelZ,HaringR,Bar-NerN, Kliger-Spatz M,Natan N,et al.M1 muscarinic agonists can modulate some of the hallmarks in Alzheimer's disease:implications in future therapy[J].J Mol Neurosci,2003,20(3):349-356.
[22]Caccamo A,Oddo S,Billings LM,Green KN, Martinez-Coria H,Fisher Aet al.M1 receptors play a central role in modulating AD-like pathology in transgenic mice[J].Neuron,2006,49(5): 671-682.
[23]Caccamo A,Fisher A,LaFerla FM.M1 agonists as a potential disease-modifying therapy for Alzheimer'sdisease[J].CurrAlzheimerRes, 2009,6(2):112-117.
[24]Rogers SL,Farlow MR,Doody RS,Mohs R, Friedhoff LT.A 24-week,double-blind,placebocontrolled trial of donepezil in patients with Alzheimer's disease.Donepezil Study Group[J].Neurology,1998,50(1):136-145.
[25]Seltzer B,Zolnouni P,Nunez M,Goldman R, Kumar D,Ieni J,et al.Efficacy of donepezil in early-stage Alzheimer disease:a randomized placebo-controlled trial[J].Arch Neurol,2004, 61(12):1852-1856.
[26]Johannsen P,Salmon E,Hampel H,Xu Y, Richardson S,Qvitzau S,et al.Assessing therapeutic efficacy in a progressive disease:a study of donepezil in Alzheimer's disease[J].CNS Drugs,2006,20(4):311-325.
[27]Winblad B,Wimo A,Engedal K,Soininen H, Verhey F,Waldemar G,et al.3-year study of donepezil therapy in Alzheimer's disease:effects of early and continuous therapy[J].Dement Geriatr Cogn Disord,2006,21(5-6):353-363.
[28]PetersenRC,ThomasRG,GrundmanM, Bennett D,Doody R,Ferris S,et al.Vitamin E and donepezil for the treatment of mild cognitive impairment[J].N Engl J Med,2005,352(23): 2379-2388.
[29]Wilcock G,Howe I,Coles H,Lilienfeld S,Truyen L, Zhu Y,et al.A long-term comparison of galantamine and donepezil in the treatment of Alzheimer's disease[J].Drugs Aging,2003,20(10): 777-789.
[30]Bullock R,Touchon J,Bergman H,Gambina G, He Y,Rapatz G,et al.Rivastigmine and donepezil treatment in moderate to moderately-severe Alzheimer's disease over a 2-year period[J].Curr Med Res Opin,2005,21(8):1317-1327.
[31]Cummings J,Lefèvre G,Small G,Appel-Dingemanse S.Pharmacokinetic rationale for the rivastigmine patch[J].Neurology,2007,69(4 Suppl 1):S10-S13.
[32]Erkinjuntti T,Skoog I,Lane R,Andrews C.Potential long-term effects of rivastigmine on disease progression may be linked to drug effects on vascular changes in Alzheimer brains[J].Int J Clin Pract,2003,57(9):756-760.
[33]Farlow MR,Doraiswamy PM,Meng X,Cooke K, Somogyi M.The effect of vascular risk factors on the efficacy of rivastigmine patch and capsule treatment in Alzheimer's disease[J].Dement Geriatr Cogn Dis Extra,2011,1(1):150-162.
[34]Birks J,McGuinness B,Craig D.Rivastigmine for vascular cognitive impairment[J].Cochrane Database Syst Rev,2013,5:CD004744.
[35]Nava-Mesa MO,Jiménez-Díaz L,Yajeya J, Navarro-Lopez JD.GABAergic neurotransmission and new strategies of neuromodulation to compensate synaptic dysfunction in early stages of Alzheimer's disease[J].Front Cell Neurosci, 2014,8:167.
[36]Reisberg B,Doody R,Stöffler A,Schmitt F, Ferris S,Möbius HJ.Memantine in moderate-tosevere Alzheimer's disease[J].N Engl J Med, 2003,348(14):1333-1341.
[37]Reisberg B,Doody R,Stöffler A,Schmitt F,Ferris S,Möbius HJ.A 24-week open-label extension study of memantine in moderate to severe Alzheimer disease[J].Arch Neurol,2006,63(1):49-54.
[38]DoodyRS,TariotPN,PfeifferE,OlinJT, Graham SM.Meta-analysis of six-month memantine trials in Alzheimer's disease[J].Alzheimers Dement,2007,3(1):7-17.
[39]SchneiderLS,DagermanKS,HigginsJP, McShane R.Lack of evidence for the efficacy of memantine in mild Alzheimer disease[J].Arch Neurol,2011,68(8):991-998.
[40]Atri A.Effective pharmacological management of Alzheimer's disease[J].Am J Manag Care, 2011,17:S346-355.
[41]Johnson JW,Kotermanski SE.Mechanism of action of memantine[J].Curr Opin Pharmacol, 2006,6(1):61-67.
[42]Giannakopoulos P,Kövari E,Gold G,von Gunten A,Hof PR,Bouras C.Pathological substrates of cognitive decline in Alzheimer's disease[J].Front Neurol Neurosci,2009,24:20-29.
[43]Rissman RA,De Blas AL,Armstrong DM.GABA(A)receptors in aging and Alzheimer's disease[J].J Neurochem,2007,103(4):1285-1292.
[44]Canas PM,Simões AP,Rodrigues RJ,Cunha RA. Predominant loss of glutamatergic terminal markers in a β-amyloid peptide model of Alzheimer's disease[J].Neuropharmacology,2014,76:51-56.
[45]Somogyi P,Klausberger T.Defined types of cortical interneurone structure space and spike timing in the hippocampus[J].J Physiol,2005, 562(Pt 1):9-26.
[46]Gong N,Li Y,Cai GQ,Niu RF,Fang Q,Wu K,et al.GABA transporter-1 activity modulates hippocampal theta oscillation and theta burst stimulation-induced long-term potentiation[J].J Neurosci,2009,29(50):15836-15845.
[47]PalopJJ,ChinJ,RobersonED,WangJ, Thwin MT,Bien-Ly N,et al.Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease[J].Neuron,2007, 55(5):697-711.
[48]Bae CY,Cho CY,Cho K,Hoon Oh B,Choi KG, Lee HS,et al.A double-blind,placebo-controlled, multicenter study of cerebrolysin for Alzheimer's disease[J].J Am Geriatr Soc,2000,48(12): 1566-1571.
[49]Wei ZH,He QB,Wang H,Su BH,Chen HZ.Metaanalysis:the efficacy of nootropic agent cerebrolysin in the treatment of Alzheimer's disease[J].J Neural Transm,2007,114(5):629-634.
[50]Froestl W,Gallagher M,Jenkins H,Madrid A, Melcher T,Teichman S,et al.SGS742:the first GABA(B)receptor antagonist in clinical trials[J].Biochem Pharmacol,2004,68(8):1479-1487.
[51]Alzforum net working for a cure.Therapeutics [EB/OL].[2015-05-01].http://www.alzforum.org/ therapeutics
[52]ClinicalTrial.gov.[EB/OL].[2015-05-01].https:// www.clinicaltrials.gov/ct2/home
[53]Cleveland DW,Hwo SY,Kirschner MW.Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly[J].J Mol Biol,1977,116(2):227-247.
[54]Reynolds MR,Reyes JF,Fu Y,Bigio EH, Guillozet-Bongaarts AL,Berry RW,et al.Tau nitration occurs at tyrosine 29 in the fibrillar lesions of Alzheimer's disease and other tauopathies[J].J Neurosci,2006,26(42):10636-10645.
[55]Cohen TJ,Guo JL,Hurtado DE,Kwong LK, Mills IP,Trojanowski JQ,et al.The acetylation of tau inhibits its function and promotes patho-logical tau aggregation[J].Nat Commun,2011, 2:252.
[56]Gong CX,Liu F,Grundke-Iqbal I,Iqbal K.Posttranslationalmodificationsoftauproteinin Alzheimer's disease[J].J Neural Transm,2005, 112(6):813-838.
[57]Luo HB,Xia YY,Shu XJ,Liu ZC,Feng Y,Liu XH,et al.SUMOylation at K340 inhibits tau degradationthroughderegulatingitsphosphorylation and ubiquitination[J].Proc Natl Acad Sci USA, 2014,111(46):16586-16591.
[58]Grundke-Iqbal I,Iqbal K,Quinlan M,Tung YC, Zaidi MS,Wisniewski HM.Microtubule-associated protein tau.A component of Alzheimer paired helical filaments[J].J Biol Chem,1986,261(13):6084-6089.
[59]Kosik KS,Joachim CL,Selkoe DJ.Microtubuleassociated protein tau(tau)is a major antigenic componentofpairedhelicalfilamentsinAlzheimer disease[J].Proc Natl Acad Sci USA,1986,83(11):4044-4048.
[60]HuYY,HeSS,WangX,DuanQH,Grundke-IqbalI, Iqbal K,et al.Levels of nonphosphorylated and phosphorylatedtauincerebrospinalfluidof Alzheimer's disease patients:an ultrasensitive bienzyme-substrate-recycleenzyme-linkedimmunosorbent assay[J].Am J Pathol,2002,160(4):1269-1278.
[61]MatsuoES,ShinRW,BillingsleyML,Van deVoorde A,O'Connor M,Trojanowski JQ,et al. Biopsy-derivedadulthumanbraintauisphosphorylated at many of the same sites as Alzheimer's disease paired helical filament tau[J].Neuron, 1994,13(4):989-1002.
[62]Gómez-Isla T,Hollister R,West H,Mui S, Growdon JH,Petersen RC,et al.Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease[J].Ann Neurol, 1997,41(1):17-24.
[63]BrundenKR,ZhangB,CarrollJ,YaoY,PotuzakJS, Hogan AM,et al.Epothilone D improves microtubule density,axonal integrity,and cognition in a transgenic mouse model of tauopathy[J].J Neurosci,2010,30(41):13861-13866.
[64]Brunden KR,Yao Y,Potuzak JS,Ferrer NI, Ballatore C,James MJ,et al.The characterization of microtubule-stabilizing drugs as possible therapeutic agents for Alzheimer's disease and related tauopathies[J].Pharmacol Res,2011, 63(4):341-351.
[65]Shiryaev N,Jouroukhin Y,Giladi E,Polyzoidou E, GrigoriadisNC,RosenmannH,etal.NAPprotects memory,increases soluble tau and reduces tau hyperphosphorylation in a tauopathy model[J].Neurobiol Dis,2009,34(2):381-388.
[66]Lee VM,Brunden KR,Hutton M,Trojanowski JQ. Developing therapeutic approaches to tau,selected kinases,and related neuronal protein targets[J].Cold Spring Harb Perspect Med,2011, 1(1):a006437.
[67]BuéeL,BussièreT,Buée-ScherrerV,DelacourteA, Hof PR.Tau protein isoforms,phosphorylation and role in neurodegenerative disorders[J].Brain Res Brain Res Rev,2000,33(1):95-130.
[68]Avila J.Tau phosphorylation and aggregation in Alzheimer's disease pathology[J].FEBS Lett, 2006,580(12):2922-2927.
[69]Alonso AC,Grundke-Iqbal I,Iqbal K.Alzheimer's diseasehyperphosphorylatedtausequesters normal tau into tangles of filaments and disassembles microtubules[J].Nat Med,1996,2(7): 783-787.
[70]Yamaguchi H,Ishiguro K,Uchida T,Takashima A, Lemere CA,Imahori K.Preferential labeling of Alzheimer neurofibrillary tangles with antisera for tauproteinkinase(TPK)I/glycogensynthase kinase-3 beta and cyclin-dependent kinase 5,a component of TPK II[J].Neuropathol, 1996,92(3):232-241.
[71]Imahori K,Uchida T.Physiology and pathology of tau protein kinases in relation to Alzheimer's disease[J].J Biochem,1997,121(2):179-188.
[72]Wen Y,Planel E,Herman M,Figueroa HY,Wang L, Liu L,et al.Interplay between cyclin-dependent kinase 5 and glycogen synthase kinase 3 beta mediated by neuregulin signaling leads to differential effects on tau phosphorylation and amyloid precursor protein processing[J].J Neurosci, 2008,28(10):2624-2632.
[73]SchneiderA,BiernatJ,vonBergenM,MandelkowE, Mandelkow EM.Phosphorylation that detaches tau protein from microtubules(Ser262,Ser214)also protects it against aggregation into Alzheimer paired helical filaments[J].Biochemistry,1999, 38(12):3549-3558.
[74]LiuF,IqbalK,Grundke-IqbalI,HartGW,GongCX. O-GlcNAcylationregulates phosphorylation of tau:a mechanism involved in Alzheimer's disease[J].Proc Natl Acad Sci USA,2004,101(29):10804-10809.
[75]MinSW,ChoSH,ZhouY,SchroederS, Haroutunian V,Seeley WW,et al.Acetylation of tau inhibits its degradation and contributes to tauopathy[J].Neuron,2010,67(6):953-966.
[76]Margittai M,Langen R.Template-assisted filament growth by parallel stacking of tau[J].Proc Natl Acad Sci USA,2004,101(28):10278-10283.
[77]Congdon EE,Kim S,Bonchak J,Songrug T, Matzavinos A,Kuret J.Nucleation-dependent tau filament formation:the importance of dimerization and an estimation of elementary rate constants[J].J Biol Chem,2008,283(20):13806-13816.
[78]Wischik CM,Edwards PC,Lai RY,Roth M, Harrington CR.Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines[J].Proc Natl Acad Sci USA,1996,93(20): 11213-11218.
[79]Crowe A,James MJ,Lee VM,Smith AB 3rd,Trojanowski JQ,Ballatore C,et al.Aminothienopyridazines and methylene blue affect Tau fibrillization via cysteine oxidation[J].J Biol Chem, 2013,288(16):11024-11037.
[80]Congdon EE,Wu JW,Myeku N,Figueroa YH, Herman M,Marinec PS,et al.Methylthioninium chloride(methylene blue)induces autophagy and attenuates tauopathyin vitroandin vivo[J].Autophagy,2012,8(4):609-622.
[81]Wen Y,Li W,Poteet EC,Xie L,Tan C,Yan LJ,et al.Alternative mitochondrial electron transfer as a novel strategy for neuroprotection[J].J Biol Chem,2011,286(18):16504-16515.
[82]Shimura H,Schwartz D,Gygi SP,Kosik KS. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival[J].J Biol Chem,2004,279(6):4869-4876.
[83]DickeyCA,YueM,LinWL,DicksonDW, Dunmore JH,Lee WC,et al.Deletion of the ubiquitin ligase CHIP leads to the accumulation,but not the aggregation,of both endogenous phospho-and caspase-3-cleaved tau species[J].J Neurosci,2006,26(26):6985-6996.
[84]Dickey CA,Dunmore J,Lu B,Wang JW,Lee WC, Kamal A,et al.HSP induction mediates selective clearance of tau phosphorylated at proline-directed Ser/Thr sites but not KXGS(MARK)sites[J].FASEB J,2006,20(6):753-755.
[85]Dickey CA,Kamal A,Lundgren K,Klosak N, Bailey RM,Dunmore J,et al.The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins[J].J Clin Invest,2007,117(3):648-658.
[86]Solit DB,Chiosis G.Development and application of Hsp90 inhibitors[J].Drug Discov Today, 2008,13(1-2):38-43.
[87]Hamano T,Gendron TF,Causevic E,Yen SH, Lin WL,Isidoro C,et al.Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression[J].Eur J Neurosci,2008,27(5):1119-1130.
[88]Delgoffe GM,Powell JD.mTOR:taking cues from the immune microenvironment[J].Immunology,2009,127(4):459-465.
[89]Harrison DE,Strong R,Sharp ZD,Nelson JF, Astle CM,Flurkey K,et al.Rapamycin fed late in life extends lifespan in genetically heterogeneous mice[J].Nature,2009,460(7253):392-395.
[90]Miller RA,Harrison DE,Astle CM,Baur JA, Boyd AR,de Cabo R,et al.Rapamycin,but not resveratrol or simvastatin,extends life span of genetically heterogeneous mice[J].J Gerontol A Biol Sci Med Sci,2011,66(2):191-201.
[91]SarkarS,FlotoRA,BergerZ,ImarisioS, CordenierA,Pasco M,et al.Lithium induces autophagy by inhibiting inositol monophosphatase[J].J Cell Biol,2005,170(7):1101-1111.
[92]Noble W,Planel E,Zehr C,Olm V,Meyerson J, Suleman F,et al.Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degenerationin vivo[J].Proc Natl Acad Sci USA,2005,102(19):6990-6995.
[93]Frost B,Jacks RL,Diamond MI.Propagation of tau misfolding from the outside to the inside of a cell[J].JBiolChem,2009,284(19):12845-12852.
[94]ClavagueraF,BolmontT,CrowtherRA, Abramowski D,Frank S,Probst A,et al.Transmission and spreading of tauopathy in transgenic mouse brain[J].Nat Cell Biol,2009,11(7): 909-913.
[95]AsuniAA,BoutajangoutA,QuartermainD, Sigurdsson EM.Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements[J].J Neurosci,2007,27(34):9115-9129.
[96]Troquier L1,Caillierez R,Burnouf S,Fernandez-Gomez FJ,Grosjean ME,Zommer N,et al.Targeting phospho-Ser422 by active Tau immuno-therapy in the THYTau22 mouse model:a suitable therapeutic approach[J].Curr Alzheimer Res,2012,9(4):397-405.
[97]RosenmannH,GrigoriadisN,KarussisD,BoimelM, Touloumi O,Ovadia H,et al.Tauopathy-like abnormalities and neurologic deficits in mice immunized with neuronal tau protein[J].Arch Neurol, 2006,63(10):1459-1467.
[98]Buchhave P,Minthon L,Zetterberg H,Wallin AK, Blennow K,Hansson O.Cerebrospinal fluid levels of β-amyloid 1-42,but not of tau,are fully changed already 5 to 10 years before the onset of Alzheimer dementia[J].Arch Gen Psychiatry, 2012,69(1):98-106.
[99]Choi SH,Kim YH,Hebisch M,Sliwinski C,Lee S, D'Avanzo C,et al.A three-dimensional human neural cell culture model of Alzheimer's disease[J].Nature,2014,515(7526):274-278.
[100]Price DL,Sisodia SS,Gandy SE.Amyloid beta amyloidosis in Alzheimer's disease[J].Curr Opin Neurol,1995,8(4):268-274.
[101]Hardy J,Selkoe DJ.The amyloid hypothesis of Alzheimer's disease:progress and problems on the road to therapeutics[J].Science,2002,297(5580):353-356.
[102]Wang H,Li R,Shen Y.β-Secretase:its biology as a therapeutic target in diseases[J].Trends Pharmacol Sci,2013,34(4):215-225.
[103]Vassar R,Bennett BD,Babu-Khan S,Kahn S, Mendiaz EA,Denis P,et al.Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE[J].Science,1999,286(5440):735-741.
[104]Yan R,Bienkowski MJ,Shuck ME,Miao H, Tory MC,Pauley AM,et al.Membrane-anchored aspartyl protease with Alzheimer's disease betasecretase activity[J].Nature,1999,402(6761): 533-537.
[105]Sinha S,Anderson JP,Barbour R,Basi GS, Caccavello R,Davis D,et al.Purification and cloning of amyloid precursor protein beta-secretase from human brain[J].Nature,1999,402(6761):537-540.
[106]Bennett BD,Babu-Khan S,Loeloff R,Louis JC, Curran E,Citron M,et al.Expression analysis of BACE2 in brain and peripheral tissues[J].J Biol Chem,2000,275(27):20647-20651.
[107]Yan R,Munzner JB,Shuck ME,Bienkowski MJ. BACE2 functions as an alternative alpha-secretase in cells[J].J Biol Chem,2001,276(36): 34019-34027.
[108]Li R,Lindholm K,Yang LB,Yue X,Citron M, Yan R,et al.Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer's disease patients[J].Proc Natl Acad Sci USA,2004,101(10):3632-3637.
[109]Yang LB,Lindholm K,Yan R,Citron M,Xia W, Yang XL,et al.Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease[J].Nat Med,2003,9(1):3-4.
[110]Seshadri S,Kamiya A,Yokota Y,Prikulis I,Kano S, Hayashi-Takagi A,et al.Disrupted-in-Schizophrenia-1 expression is regulated by beta-site amyloid precursor protein cleaving enzyme-1-neuregulin cascade[J].Proc Natl Acad Sci USA,2010,107(12):5622-5627.
[111]He P,Zhong Z,Lindholm K,Berning L,Lee W, Lemere C,et al.Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer's mice[J].J Cell Biol,2007,178(5):829-841.
[112]Sun X,He G,Qing H,Zhou W,Dobie F,Cai F,et al.Hypoxia facilitates Alzheimer's disease pathogenesis by up-regulating BACE1 gene expression[J].Proc Natl Acad Sci USA,2006, 103(49):18727-18732.
[113]Sastre M,Dewachter I,Landreth GE,Willson TM, Klockgether T,van Leuven F,et al.Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase[J].J Neurosci,2003,23(30):9796-9804.
[114]Zhang X,Zhou K,Wang R,Cui J,Lipton SA, Liao FF,et al.Hypoxia-inducible factor 1alpha(HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation[J].J Biol Chem,2007,282(15):10873-10880.
[115]Huse JT,Pijak DS,Leslie GJ,Lee VM,Doms RW. Maturation and endosomal targeting of beta-site amyloid precursor protein-cleaving enzyme.The Alzheimer's disease beta-secretase[J].J Biol Chem,2000,275(43):33729-33737.
[116]Haniu M,Denis P,Young Y,Mendiaz EA,Fuller J, Hui JO,et al.Characterization of Alzheimer's beta-secretase protein BACE.A pepsin family memberwithunusualproperties[J].JBiol Chem,2000,275(28):21099-21106.
[117]Shiba T,Kametaka S,Kawasaki M,Shibata M,Waguri S,Uchiyama Y,et al.Insights into the phosphoregulationofbeta-secretasesorting signal by the VHS domain of GGA1[J].Traffic, 2004,5(6):437-448.
[118]Vetrivel KS,Meckler X,Chen Y,Nguyen PD, Seidah NG,Vassar R,et al.Alzheimer disease Abeta production in the absence of S-palmitoylation-dependent targeting of BACE1 to lipid rafts[J].J Biol Chem,2009,284(6):3793-3803.
[119]Kang EL,Cameron AN,Piazza F,Walker KR, Tesco G.Ubiquitin regulates GGA3-mediated degradation of BACE1[J].J Biol Chem,2010, 285(31):24108-24119.
[120]Costantini C,Ko MH,Jonas MC,Puglielli L.A reversible form of lysine acetylation in the ER and Golgi lumen controls the molecular stabilization of BACE1[J].Biochem J,2007,407(3):383-395.
[121]Kimura R,Devi L,Ohno M.Partial reduction of BACE1 improves synaptic plasticity,recent and remote memories in Alzheimer's disease transgenic mice[J].J Neurochem,2010,113(1): 248-261.
[122]Filser S,Ovsepian SV,Masana M,Blazquez-Llorca L,Brandt Elvang A,Volbracht C,et al. Pharmacological inhibition of BACE1 impairs synaptic plasticity and cognitive functions[J].Biol Psychiatry,2015,77(8):729-739.
[123]Cole SL,Vassar R.The Alzheimer's disease betasecretase enzyme,BACE1[J].Mol Neurodegener,2007,2:22.
[124]GhoshAK,BrindisiM,TangJ.Developing β-secretase inhibitors for treatment of Alzheimer's disease[J].J Neurochem,2011,120:71-83.
[125]Luo X,Yan R.Inhibition of BACE1 for therapeutic use in Alzheimer's disease[J].Int J Clin Exp Pathol,2010,3(6):618-628.
[126]Edbauer D,Winkler E,Regula JT,Pesold B, Steiner H,Haass C.Reconstitution of gammasecretase activity[J].Nat Cell Biol,2003,5(5): 486-488.
[127]FrancisR,McGrathG,ZhangJ,RuddyDA,SymM, Apfeld J,et al.aph-1 and pen-2 are required for Notchpathwaysignaling,gamma-secretasecleavage of betaAPP,and presenilin protein accumulation[J].Dev Cell,2002,3(1):85-97.
[128]Wolfe MS,Xia W,Ostaszewski BL,Diehl TS, Kimberly WT,Selkoe DJ.Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity[J].Nature,1999,398(6727):513-517.
[129]Xia W,Zhang J,Perez R,Koo EH,Selkoe DJ.Interaction between amyloid precursor protein and presenilins in mammalian cells:implications for the pathogenesis of Alzheimer disease[J].Proc Natl Acad Sci USA,1997,94(15):8208-8213.
[130]Selkoe DJ.Presenilin,Notch,and the genesis and treatment of Alzheimer's disease[J].Proc Natl Acad Sci USA,2001,98(20):11039-11041.
[131]Selkoe DJ,Wolfe MS.Presenilin:running with scissors in the membrane[J].Cell,2007,131(2):215-221.
[132]Artavanis-Tsakonas S,Rand MD,Lake RJ. Notch signaling:cell fate control and signal integration in development[J].Science,1999,284(5415):770-776.
[133]Doody RS,Raman R,Farlow M,Iwatsubo T, Vellas B,Joffe S,et al.A phase 3 trial of semagacestat for treatment of Alzheimer's disease[J].N Engl J Med,2013,369(4):341-350.
[134]Bush AI,Tanzi RE.Therapeutics for Alzheimer's disease based on the metal hypothesis[J].Neurotherapeutics,2008,5(3):421-432.
[135]Faller P,Hureau C,Berthoumieu O.Role of metal ions in the self-assembly of the Alzheimer's amyloid-β peptide[J].Inorg Chem,2013,52(21): 12193-12206.
[136]Crouch PJ,Savva MS,Hung LW,Donnelly PS, Mot AI,Parker SJ,et al.The Alzheimer's therapeutic PBT2 promotes amyloid-β degradation and GSK3 phosphorylation via a metal chaperone activity[J].J Neurochem,2011,119(1): 220-230.
[137]MawuenyegaKG,SigurdsonW,OvodV, Munsell L,Kasten T,Morris JC,et al.Decreased clearance of CNS beta-amyloid in Alzheimer's disease[J].Science,2010,330(6012):1774.
[138]Suh YH,Checler F.Amyloid precursor protein, presenilins,andalpha-synuclein:molecular pathogenesis and pharmacological applications in Alzheimer's disease[J].Pharmacol Rev, 2002,54(3):469-525.
[139]Turner AJ,Fisk L,Nalivaeva NN.Targeting amyloid-degrading enzymes as therapeutic strategies in neurodegeneration[J].Ann N Y Acad Sci,2004,1035:1-20.
[140]Schenk D,Barbour R,Dunn W,Gordon G, Grajeda H,Guido T,et al.Immunization with amyloid-beta attenuates Alzheimer-disease-likepathology in the PDAPP mouse[J].Nature, 1999,400(6740):173-177.
[141]BardF,CannonC,BarbourR,BurkeRL, Games D,Grajeda H,et al.Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease[J].Nat Med,2000,6(8):916-919.
[142]Janus C,Pearson J,McLaurin J,Mathews PM, Jiang Y,Schmidt SD,et al.A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease[J].Nature,2000,408(6815):979-982.
[143]Morgan D,Diamond DM,Gottschall PE,Ugen KE, Dickey C,Hardy J,et al.A beta peptide vaccination prevents memory loss in an animal model of Alzheimer'sdisease[J].Nature,2000,408(6815):982-985.
[144]Hartman RE,Izumi Y,Bales KR,Paul SM, Wozniak DF,Holtzman DM.Treatment with an amyloid-beta antibody ameliorates plaque load, learning deficits,and hippocampal long-term potentiation in a mouse model of Alzheimer's disease[J].J Neurosci,2005,25(26):6213-6220.
[145]KotilinekLA,BacskaiB,WestermanM, Kawarabayashi T,Younkin L,Hyman BT,et al. Reversible memory loss in a mouse transgenic model of Alzheimer's disease[J].J Neurosci, 2002,22(15):6331-6335.
[146]Wilcock DM,Rojiani A,Rosenthal A,Levkowitz G, Subbarao S,Alamed J,et al.Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition[J].J Neurosci,2004,24(27):6144-6151.
[147]Gandy S,DeMattos RB,Lemere CA,Heppner FL, Leverone J,Aguzzi A,et al.Alzheimer's Abeta vaccination of rhesus monkeys(Macaca mulatta)[J].Mech Ageing Dev,2004,125(2):149-151.
[148]LemereCA,BeierschmittA,IglesiasM, Spooner ET,Bloom JK,Leverone JF,et al. Alzheimer's disease abeta vaccine reduces central nervous system abeta levels in a non-human primate,the Caribbean vervet[J].Am J Pathol, 2004,165(1):283-297.
[149]Orgogozo JM,Gilman S,Dartigues JF,Laurent B, Puel M,Kirby LC,et al.Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization[J].Neurology,2003,61(1):46-54.
[150]GilmanS,KollerM,BlackRS,JenkinsL,GriffithSG, Fox NC,et al.Clinical effects of Abeta immunization(AN1792)in patients with AD in an interrupted trial[J].Neurology,2005,64(9):1553-1562.
[151]Cribbs DH,Ghochikyan A,Vasilevko V,Tran M, Petrushina I,Sadzikava N,et al.Adjuvant-dependent modulation of Th1 and Th2 responses to immunization with beta-amyloid[J].Int Immunol, 2003,15(4):505-514.
[152]Nicoll JA,Wilkinson D,Holmes C,Steart P, MarkhamH,WellerRO.Neuropathologyofhuman Alzheimer disease after immunization with amyloid-beta peptide:a case report[J].Nat Med, 2003,9(4):448-452.
[153]Ferrer I,Boada Rovira M,Sánchez Guerra ML, Rey MJ,Costa-Jussá F.Neuropathology and pathogenesis of encephalitis following amyloidbeta immunization in Alzheimer's disease[J].Brain Pathol,2004,14(1):11-20.
[154]Town T,Vendrame M,Patel A,Poetter D,Delle-Donne A,Mori T,et al.Reduced Th1 and enhanced Th2 immunity after immunization with Alzheimer's beta-amyloid(1-42)[J].J Neuroimmunol,2002,132(1-2):49-59.
[155]Monsonego A,Zota V,Karni A,Krieger JI, Bar-Or A,Bitan G,et al.Increased T cell reactivity to amyloid beta protein in older humans and patients with Alzheimer disease[J].J Clin Invest, 2003,112(3):415-422.
[156]Bard F,Barbour R,Cannon C,Carretto R,Fox M, Games D,et al.Epitope and isotype specificities of antibodies to beta-amyloid peptide for protection against Alzheimer's disease-like neuropathology[J].Proc Natl Acad Sci USA,2003,100(4):2023-2028.
[157]McLaurin J,Cecal R,Kierstead ME,Tian X, Phinney AL,Manea M,et al.Therapeutically effective antibodies against amyloid-beta peptide target amyloid-beta residues 4-10 and inhibit cytotoxicity and fibrillogenesis[J].Nat Med, 2002,8(11):1263-1269.
[158]Seabrook TJ,Bloom JK,Iglesias M,Spooner ET, Walsh DM,Lemere CA.Species-specific immune response to immunization with humanversusrodent A beta peptide[J].Neurobiol Aging, 2004,25(9):1141-1151.
[159]Seabrook TJ,Thomas K,Jiang L,Bloom J, Spooner E,Maier M,et al.Dendrimeric Abeta1-15 is an effective immunogen in wildtype and APP-tg mice[J].Neurobiol Aging,2007,28(6): 813-823.
[160]Bohrmann B,Baumann K,Benz J,Gerber F, Huber W,Knoflach F,et al.Gantenerumab:a novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cellmediated removal of human amyloid-β[J].J Alzheimers Dis,2012,28(1):49-69.
[161]Wyss-Coray T,Mucke L.Inflammation in neurodegenerative disease-a double-edged sword[J].Neuron,2002,35(3):419-432.
[162]Perry VH.Contribution of systemic inflammation to chronic neurodegeneration[J].Acta Neuropathol,2010,120(3):277-286.
[163]Kim SH,Carney DF,Hammer CH,Shin ML. Nucleated cell killing by complement:effects of C5b-9 channel size and extracellular Ca2+on the lytic process[J].J Immunol,1987,138(5): 1530-1536.
[164]Rogers J,Cooper NR,Webster S,Schultz J, McGeer PL,Styren SD,et al.Complement activation by beta-amyloid in Alzheimer disease[J].Proc Natl Acad Sci USA,1992,89(21):10016-10020.
[165]Yang LB,Li R,Meri S,Rogers J,Shen Y.Deficiency of complement defense protein CD59 may contribute to neurodegeneration in Alzheimer's disease[J].J Neurosci,2000,15(20): 7505-7509.
[166]Yang L,Lindholm K,Konishi Y,Li R,Shen Y. Target depletion of distinct tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival through different signal transduction pathways[J].J Neurosci,2002,22(8): 3025-3032.
[167]Shen Y,Yang L,Li R.What does complement do in Alzheimer's disease?Old molecules with new insights[J].Transl Neurodegener,2013,2(1):21.
[168]Lian H,Yang L,Lu H,Lian H,Yang L,Cole A,et al.NF-kB-Activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease[J].Neuron,2015,85(1):101-115.
[169]Krstic D,Knuesel I.Deciphering the mechanism underlying late-onset Alzheimer disease[J].Nat Rev Neurol,2013,9(1):25-34.
[170]McGeer PL,McGeer EG.The amyloid cascadeinflammatory hypothesis of Alzheimer disease: implications for therapy[J].Acta Neuropathol, 2013,126(4):479-497.
[171]Lawson LJ,Perry VH,Gordon S.Turnover of resident microglia in the normal adult mouse brain[J].Neuroscience,1992,48(2):405-415.
[172]Gehrmann J,Matsumoto Y,Kreutzberg GW. Microglia:intrinsic immuneffector cell of the brain[J].Brain Res Brain Res Rev,1995,20(3): 269-287.
[173]Akiyama H,Barger S,Barnum S,Bradt B, Bauer J,Cole GM,et al.Inflammation and Alzheimer's disease[J].Neurobiol Aging,2000, 21(3):383-421.
[174]HenekaMT,CarsonMJ,ElKhouryJ,LandrethGE, Brosseron F,Feinstein DL,et al.Neuroinflammation in Alzheimer's disease[J].Lancet Neurol, 2015,14(4):388-405.
[175]Tuppo EE,Arias HR.The role of inflammation in Alzheimer's disease[J].Int J Biochem Cell Biol, 2005,37(2):289-305.
[176]Paresce DM,Chung H,Maxfield FR.Slow degradation of aggregates of the Alzheimer's disease amyloid beta-protein by microglial cells[J].J Biol Chem,1997,272(46):29390-29397.
[177]Chung H,Brazil MI,Soe TT,Maxfield FR.Uptake, degradation,and release of fibrillar and soluble forms of Alzheimer's amyloid beta-peptide by microglial cells[J].J Biol Chem,1999,274(45):32301-32308.
[178]Majumdar A,Chung H,Dolios G,Wang R, Asamoah N,Lobel P,et al.Degradation of fibrillar forms of Alzheimer's amyloid beta-peptide by macrophages[J].Neurobiol Aging,2008,29(5):707-715.
[179]Chesneau V,Vekrellis K,Rosner MR,Selkoe DJ. Purified recombinant insulin-degrading enzyme degrades amyloid beta-protein but does not promote its oligomerization[J].Biochem J,2000, 351(Pt 2):509-516.
[180]Tamboli IY,Barth E,Christian L,Siepmann M, Kumar S,Singh S,et al.Statins promote the degradationofextracellularamyloid{beta}-peptide by microglia via stimulation of exosomeassociated insulin-degrading enzyme(IDE)secretion[J].J Biol Chem,2010,285(48): 37405-37414.
[181]Qiu WQ,Walsh DM,Ye Z,Vekrellis K,Zhang J, Podlisny MB,et al.Insulin-degrading enzyme regulates extracellular levels of amyloid betaprotein by degradation[J].J Biol Chem,1998,273(49):32730-32738.
[182]Colonna M.TREMs in the immune system and beyond[J].Nat Rev Immunol,2003,3(6):445-453.
[183]Wang Y,Cella M,Mallinson K,Ulrich JD,Young KL,Robinette ML,et al.TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model[J].Cell,2015,160(6): 1061-1071.
[184]Jay TR,Miller CM,Cheng PJ,Graham LC, Bemiller S,Broihier ML,et al.TREM2 deficiency eliminates TREM2+inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models[J].J Exp Med,2015,212(3):278-295.
[185]Guerreiro R,Wojtas A,Bras J,Carrasquillo M, Rogaeva E,Majounie E,et al.TREM2 variants in Alzheimer's disease[J].N Engl J Med,2013, 368(2):117-127.
[186]Neumann H,Daly MJ.Variant TREM2 as risk factor for Alzheimer's disease[J].N Engl J Med,2013,368(2):182-184.
[187]JonssonT,StefanssonH,SteinbergS, Jonsdottir I,Jonsson PV,Snaedal J,et al.Variant of TREM2 associated with the risk of Alzheimer's disease[J].N Engl J Med,2013,368(2):107-116.
[188]LeinengaG,GötzJ.Scanningultrasoundremoves amyloid-β and restores memory in an Alzheimer's disease mouse model[J].Sci Transl Med, 2015,7(278):278ra33
[189]Hickman SE,Allison EK,El Khoury J.Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice[J].J Neurosci,2008,28(33):8354-8360.
[190]Krabbe G,Halle A,Matyash V,Rinnenthal JL, Eom GD,Bernhardt U,et al.Functional impairment of microglia coincides with beta-amyloid deposition in mice with Alzheimer-like pathology[J].PLoS One,2013,8(4):e60921.
[191]Edison P,Archer HA,Gerhard A,Hinz R, Pavese N,Turkheimer FE,et al.Microglia,amyloid,and cognition in Alzheimer's disease:An [11C](R)PK11195-PET and[11C]PIB-PET study[J].Neurobiol Dis,2008,32(3):412-419.
[192]Fuhrmann M,Bittner T,Jung CK,Burgold S, PageRM,MittereggerG,etal.Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease[J].Nat Neurosci,2010,13(4):411-413.
[193]LeeS,VarvelNH,KonerthME,XuG,CardonaAE, Ransohoff RM,et al.CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models[J].Am J Pathol,2010,177(5):2549-2562.
[194]Balducci C,Mehdawy B,Mare L,Giuliani A, Lorenzini L,Sivilia S,et al.The γ-secretase modulator CHF5074 restores memory and hippocampalsynapticplasticityinplaque-freeTg2576 mice[J].J Alzheimers Dis,2011,24(4):799-816.
[195]Santello M,Volterra A.Synaptic modulation by astrocytes via Ca2+-dependent glutamate release[J].Neuroscience,2009,158(1):253-259.
[196]Walz W.Role of astrocytes in the clearance of excess extracellular potassium[J].Neurochem Int,2000,36(4-5):291-300.
[197]Turner DA,Adamson DC.Neuronal-astrocyte metabolic interactions:understanding the transition into abnormal astrocytoma metabolism[J].J Neuropathol Exp Neurol,2011,70(3):167-176.
[198]Bélanger M,Allaman I,Magistretti PJ.Brain energy metabolism:focus on astrocyte-neuron metabolic cooperation[J].Cell Metab,2011,14(6):724-738.
[199]RodríguezJJ,OlabarriaM,ChvatalA,Verkhratsky A. Astroglia in dementia and Alzheimer's disease[J].Cell Death Differ,2009,16(3):378-385.
[200]Wyss-Coray T,Loike JD,Brionne TC,Lu E, Anankov R,Yan F,et al.Adult mouse astrocytes degrade amyloid-betain vitroandin situ[J].Nat Med,2003,9(4):453-457.
[201]Koistinaho M,Lin S,Wu X,Esterman M,Koger D, Hanson J,et al.Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides[J].Nat Med,2004,10(7):719-726.
[202]Kraft AW,Hu X,Yoon H,Yan P,Xiao Q,Wang Y,et al.Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice[J].FASEB J,2013,27(1):187-198.
[203]Aguirre-Rueda D,Guerra-Ojeda S,Aldasoro M, Iradi A,Obrador E,Ortega A,et al.Astrocytes protect neurons from Aβ1-42 peptide-induced neurotoxicity increasing TFAM and PGC-1 and decreasing PPAR-γ and SIRT-1[J].Int J Med Sci,2015,12(1):48-56.
[204]Jo S,Yarishkin O,Hwang YJ,Chun YE,Park M,Woo DH,et al.GABA from reactive astrocytes impairs memory in mouse models of Alzheimer's disease[J].Nat Med,2014,20(8):886-896.
[205]Furman JL,Sama DM,Gant JC,Beckett TL, Murphy MP,Bachstetter AD,et al.Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer's disease[J].J Neurosci,2012,32(46):16129-16140.
[206]Shen Y,Meri S.Yin and Yang:complement activation and regulation in Alzheimer's disease[J].Prog Neurobiol,2003,70(6):463-472.
[207]Shen Y,Sullivan T,Lee CM,Meri S,Shiosaki K, Lin CW.Induced expression of neuronal membrane attack complex and cell death by Alzheimer's beta-amyloid peptide[J].Brain Res,1998, 796(1-2):187-197.
[208]Shen Y,Li R,McGeer EG,McGeer PL.Neuronal expression of mRNAs for complement proteins of the classical pathway in Alzheimer brain[J].Brain Res,1997,769(2):391-395.
[209]Ho L,Pieroni C,Winger D,Purohit DP,Aisen PS, Pasinetti GM.Regional distribution of cyclooxygenase-2inthehippocampalformationin Alzheimer's disease[J].J Neurosci Res,1999, 57(3):295-303.
[210]Du Yan S,Zhu H,Fu J,Yan SF,Roher A, Tourtellotte WW,et al.Amyloid-beta peptide-receptor for advanced glycation endproduct interactionelicitsneuronalexpressionofmacrophage-colony stimulating factor:a proinflammatory pathway in Alzheimer disease[J].Proc Natl Acad Sci USA,1997,94(10):5296-5301.
[211]Xanthos DN,Sandkühler J.Neurogenic neuroinflammation:inflammatory CNS reactions in response to neuronal activity[J].Nat Rev Neurosci,2014,15(1):43-53.
[212]Wyss-CorayT.InflammationinAlzheimerdisease: driving force,bystander or beneficial response?[J].Nat Med,2006,12(9):1005-1015.
[213]Stellwagen D,Malenka RC.Synaptic scaling mediated by glial TNF-alpha[J].Nature,2006,440(7087):1054-1059.
[214]Wahl SM.Transforming growth factor beta(TGF-beta)in inflammation:a cause and a cure[J].J Clin Immunol,1992,12(2):61-74.
[215]Bruns H,Meinken C,Schauenberg P,Härter G, Kern P,Modlin RL,et al.Anti-TNF immunotherapy reduces CD8+T cell-mediated antimicrobial activity againstMycobacterium tuberculosisin humans[J].J Clin Invest,2009,119(5):1167-1177.
[216]Magro F,Portela F.Management of inflammatory bowel disease with infliximab and other anti-tumor necrosis factor alpha therapies[J].BioDrugs, 2010,24(Suppl 1):3-14.
[217]Cheng X,Yang L,He P,Li R,Shen Y.Differential activation of tumor necrosis factor receptors distinguishes between brains from Alzheimer's disease and non-demented patients[J].J Alzheimers Dis,2010,19(2):621-630.
[218]Yang L,Lindholm K,Konishi Y,Li R,Shen Y. Target depletion of distinct tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival through different signal transduction pathways[J].J Neurosci,2002,22(8): 3025-3032.
[219]Li R,Yang L,Lindholm K,Konishi Y,Yue X, Hampel H,et al.Tumor necrosis factor death receptor signaling cascade is required for amyloid-beta protein-induced neuron death[J].J Neurosci,2004,24(7):1760-1771.
[220]Cheng X,Shen Y,Li R.Targeting TNF:a therapeutic strategy for Alzheimer's disease[J].Drug Discov Today,2014,19(11):1822-1827.
[221]Jiang H,He P,Xie J,Staufenbiel M,Li R,Shen Y. Genetic deletion of TNFRII gene enhances the Alzheimer-like pathology in an APP transgenic mouse model via reduction of phosphorylated IκBα[J].Hum Mol Genet,2014,23(18):4906-4918.
[222]Birch AM,Katsouri L,Sastre M.Modulation of inflammation in transgenic models of Alzheimer's disease[J].J Neuroinflammation,2014,11:25.
[223]Kiyota T,Okuyama S,Swan RJ,Jacobsen MT, Gendelman HE,Ikezu T.CNS expression of antiinflammatory cytokine interleukin-4 attenuates Alzheimer's disease-like pathogenesis in APP+PS1 bigenic mice[J].FASEB J,2010,24(8): 3093-3102.
[224]Guillot-SestierMV,DotyKR,GateD,RodriguezJJr, Leung BP,Rezai-Zadeh K,et al.IL-10 deficiency rebalances innate immunity to mitigate Alzheimerlike pathology[J].Neuron,2015,85(3):534-548.
[225]Town T,Laouar Y,Pittenger C,Mori T,Szekely CA, Tan J,et al.Blocking TGF-beta-Smad2/3 innate immunesignalingmitigatesAlzheimer-like pathology[J].Nat Med,2008,14(6):681-687.
[226]Chakrabarty P,Li A,Ceballos-Diaz C,Eddy JA, Funk CC,Moore B,et al.IL-10 alters immuno-proteostasis in APP mice,increasing plaque burden and worsening cognitive behavior[J].Neuron,2015,85(3):519-533.
[227]Chakrabarty P,Ceballos-Diaz C,Beccard A, Janus C,Dickson D,Golde TE,et al.IFN-gamma promotes complement expression and attenuates amyloid plaque deposition in amyloid beta precursor protein transgenic mice[J].J Immunol,2010,184(9):5333-5343.
[228]ChakrabartyP,HerringA,Ceballos-DiazC,DasP, Golde TE.Hippocampal expression of murine TNFα results in attenuation of amyloid depositionin vivo[J].Mol Neurodegener,2011,6:16.
[229]Chakrabarty P,Jansen-West K,Beccard A, Ceballos-Diaz C,Levites Y,Verbeeck C,et al. Massive gliosis induced by interleukin-6 suppressesAbetadepositioninvivo:evidenceagainst inflammation as a driving force for amyloid deposition[J].FASEB J,2010,24(2):548-559.
[230]Matousek SB,Ghosh S,Shaftel SS,Kyrkanides S, Olschowka JA,O'Banion MK.Chronic IL-1βmediated neuroinflammation mitigates amyloid pathology in a mouse model of Alzheimer's disease without inducing overt neurodegeneration[J].J Neuroimmune Pharmacol,2012,7(1):156-164.
[231]Vom Berg J,Prokop S,Miller KR,Obst J,Kälin RE, Lopategui-Cabezas I,et al.Inhibition of IL-12/ IL-23 signaling reduces Alzheimer's disease-like pathology and cognitive decline[J].Nat Med, 2012,18(12):1812-1819.
[232]Reid KB,Porter RR.The proteolytic activation systems of complement[J].Annu Rev Biochem, 1981,50:433-464.
[233]Müller-Eberhard HJ.Molecular organization and function of the complement system[J].Annu Rev Biochem,1988,57:321-347.
[234]Peitsch MC,Tschopp J.Assembly of macromolecular pores by immune defense systems[J].Curr Opin Cell Biol,1991,3(4):710-716.
[235]Holers VM.Complement and its receptors:New insights into human disease[J].Annu Rev Immunol,2014,32:433-459.
[236]Rogers J,Shen Y.A perspective on inflammation in Alzheimer's disease[J].Ann N Y Acad Sci,2000,924:132-135.
[237]Wyss-CorayT,RogersJ.Inflammationin Alzheimer disease-a brief review of the basic science and clinical literature[J].Cold SpringHarb Perspect Med,2012,2(1):a006346.
[238]Obermann KR,Morris JC,Roe CM.Exploration of 100 commonly used drugs and supplements on cognition in older adults[J].Alzheimers Dement,2013,9(6):724-732.
[239]in't Veld BA,Launer LJ,Hoes AW,Ott A,Hofman A,Breteler MM,et al.NSAIDs and incident Alzheimer's disease.The Rotterdam study[J].Neurobiol Aging,1998,19(6):607-611.
[240]in t'Veld BA,Ruitenberg A,Hofman A,Launer LJ, van Duijn CM,Stijnen T,et al.Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease[J].N Engl J Med,2001,345(21): 1515-1521.
[241]Lim GP,Yang F,Chu T,Chen P,Beech W, Teter B,et al.Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease[J].J Neurosci,2000,20(15):5709-5714.
[242]Lleó A,Berezovska O,Herl L,Raju S,Deng A, Bacskai BJ,et al.Nonsteroidal anti-inflammatory drugs lower Abeta42 and change presenilin 1 conformation[J].Nat Med,2004,10(10):1065-1066.
[243]Small GW,Siddarth P,Silverman DH,Ercoli LM, Miller KJ,Lavretsky H,et al.Cognitive and cerebral metabolic effects of celecoxibversusplacebo in people with age-related memory loss:randomized controlled study[J].Am J Geriatr Psychiatry,2008,16(12):999-1009.
[244]Boyd TD,Bennett SP,Mori T,Governatori N, Runfeldt M,Norden M,et al.GM-CSF upregulated in rheumatoid arthritis reverses cognitive impairment and amyloidosis in Alzheimer mice[J].J Alzheimers Dis,2010,21(2):507-518.
[245]He P,Cheng X,Staufenbiel M,Li R,Shen Y. Long-term treatment of thalidomide ameliorates amyloid-likepathologythroughinhibitionof β-secretase in a mouse model of Alzheimer's disease[J].PLoS One,2013,8(2):e55091.
[246]Phiel CJ,Wilson CA,Lee VM,Klein PS.GSK-3alpharegulatesproductionofAlzheimer's diseaseamyloid-betapeptides[J].Nature, 2003,423(6938):435-439.
[247]Genin E,Hannequin D,Wallon D,Sleegers K, Hiltunen M,Combarros O,et al.APOE and Alzheimer disease:a major gene with semidominant inheritance[J].Mol Psychiatry,2011, 16(9):903-907.
Development of potential therapeutic targets of and approaches to Alzheimer disease
BI Dan-lei1,WEN Lang1,XIONG Wei2,SHEN Yong1
(1.Neurodegenerative Disease Research Center,2.Laboratory for Integrative Neuroscience, School of Life Sciences,Chinese Academy of Sciences Key Laboratory of Brain Function and Disease, The Chemistry of Life Collaborative Innovation Center,University of Science and Technology of China,Hefei 230027,China)
Alzheimer disease(AD)is a neurodegenerative disease mainly seen in elder populations aged 65 or above.The pathological hallmarks in the AD brain include senile plaques due to amyloid-β(Aβ)deposition,neurofibrils composed of hyperphosphorylated tau within neurons,chronic inflammation and neuron death,which,besides aging,are regarded as the main pathogenesis of AD. Here we review the current development of therapeutic approaches to AD.All the five AD therapeutics approved so far are designed to improve AD patient cognition by decreasing the effects of excitatory neurotransmitters,the efficiency of which,however,is quite limited.Moreover,the targets of most potential drugs acting on the nervous system are the excitatory system,but there is little progress.As for drug development based on the tau hypothesis,the main strategies are to inhibit tau phosphorylation,fibrillization and transmission.However,due to the difficulties in specifically inhibiting tau phosphorylation,two of the four tau drugs in clinical trials are tau active vaccines,which show no promise.In the Aβ hypothesis,the main strategies are to inhibit Aβ production/aggregation and promote Aβ clearance.Due to the severe adverse effects of γ-secretase inhibitors,the main approaches are to develop β-secretase(BACE1)inhibitors and Aβ vaccines.In addition,another potential therapeutic approach is to inhibit chronic inflammation in the AD brain.None of the nonsteroidal antiinflammatory drugs(NSAIDs)have succeeded.Potential antiinflammatory drugs acting on other inflammation factors,such as TNF and TGF,are still in clinical trials and making good progress.Generally speaking, the major obstacle to AD drug development is that the potential molecules lack druggability,and that most of the clinical trials have failed due to adverse effects and insufficiency.However,there are 82 potential drugs in clinical trails including 18 currently in phaseⅢ/Ⅳ.Meanwhile,application of new techniques,such as computer designed precise-targeting,and CRISPR/caspase9,is expected to significantly accelerate AD drug discovery.
neurodegenerative disease;Alzheimer disease;drug discovery;therapeutic uses;amyloid-beta-peptides;tau protein;inflammation
The project supported by International Postdoctoral Exchange Fellowship Program;and Recruitment Program of Global Experts and the Research Foundation of the University of Science and Technology of China
SHEN Yong,E-mail:yongshen@ustc.edu.cn

R966,R971
A
1000-3002-(2015)04-0507-30
10.3867/j.issn.1000-3002.2015.04.001
2015-05-20接受日期:2015-07-08)
(本文编辑:乔虹)
博士后国际交流计划资助;中国科技大学及国家千人计划学者启动基金资助。
申勇,E-mail:yongshen@ustc.edu.cn
猜你喜欢
波谱学杂志(2022年1期)2022-03-15
海外星云(2021年9期)2021-10-14
现代仪器与医疗(2021年1期)2021-06-09
神经损伤与功能重建(2020年10期)2020-12-23
神经损伤与功能重建(2020年11期)2020-12-01
大众健康(2020年7期)2020-08-25
天津医科大学学报(2019年6期)2019-08-13
分析化学(2017年12期)2017-12-25
安徽医科大学学报(2015年9期)2015-12-16
飞碟探索(2015年11期)2015-09-10
