
2015 年先天性心脏病相关性肺动脉高压诊治中国专家共识
2015-07-11 03:11:24中国医师协会心血管内科医师分会
中国介入心脏病学杂志 2015年2期
中国医师协会心血管内科医师分会
先天性心脏病(congenital heart disease,CHD)是我国引起肺动脉高压(pulmonary arterial hypertension,PAH)最常见的原因之一,诸多患者因PAH 而失去手术机会。然而,CHD相关性PAH(PAH associated with CHD,PAH-CHD)如何诊治,目前尚无统一标准。为使患者获得最佳治疗方案,特撰写此共识,以期为PAH-CHD 规范化诊治提供借鉴。
1 PAH-CHD 的定义
PAH-CHD 是指由体-肺分流型CHD 所引起的肺动脉压(pulmonary artery pressure,PAP)升高,系毛细血管前型肺高压(pulmonary hypertension,PH)的一种,诊断标准与其他类型PAH 相同。
2 PAH-CHD 的发生率
PAH-CHD 患病率约(1.6 ~12.5)/106,成人CHD 患者有5% ~10%将出现PAH[1-3]。上世纪50 年代欧洲艾森曼格综合征(Eisenmenger's syndrome,ES)发生率为8%,现降至4%[3]。我国目前尚无相关流行病学资料。
3 PAH-CHD 的临床分类、分级与分期
3.1 PAH-CHD 的临床分类
PAH-CHD 临床分为ES、PAH 合并体-肺分流、PAH 合并小型CHD 和术后PAH 四类(表1)。
3.2 PAH-CHD 的分级
与其他类型PAH 不同,在PAH-CHD 患者中,肺/体循环压力比值(pulmonary-to-systemic pressure ratio,Pp/Ps)和阻力比值(pulmonary-to-systemic vascular resistance ratio,Rp/Rs)更能反映PAH-CHD 严重程度。PAH-CHD 的分级见表2。
3.3 PAH-CHD 的分期
根据体-肺分流程度,将PAH-CHD 分为动力型和阻力型二期。 (1)动力型PAH 期:患者存在PAH,但肺血管尚未发生严重病变,关闭缺损之后PAP 可降至正常。(2)阻力型PAH 期:肺血管已发生不可逆病变,关闭缺损后,患者PAP不能降至正常,或反而升高而出现术后持续性PAH。

表1 先天性心脏病相关性肺动脉高压临床分类
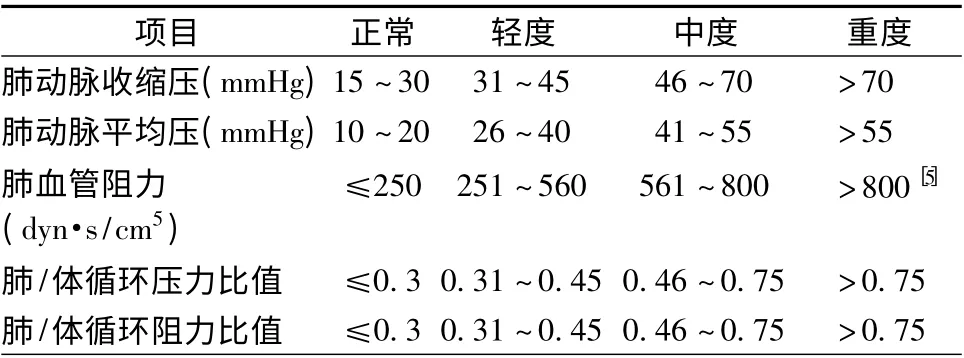
表2 先天性心脏病相关性肺动脉高压分级
如何将动力型和阻力型PAH 完全分开,目前尚无统一标准。根据2010 年欧洲心脏病学会(ESC)成人CHD 管理指南[6],仍以肺/体循环血流量比值(Qp/Qs)>1.5 作为区分动力型和阻力型PAH 标准,即PAP 显著升高同时Qp/Qs<1.5 提示患者已进入阻力型PAH 期。
4 PAH-CHD 的预后
PAH-CHD 预后通常针对ES 而言,其3 年生存率约77%,平均寿命(32.5 ±16)岁,30 岁、40 岁和50 岁的存活率分别为75%、70%和50%[7]。常见并发症有栓塞、出血、肺动脉血栓形成、红细胞增多症、感染、心律失常、猝死、肝肾功能异常和骨骼疾病等。主要死亡原因为猝死(29.5%)、心力衰竭(22.9%)和咯血(11.4%)[8]。
5 PAH-CHD 的临床表现
5.1 PAH-CHD 的症状
PAH-CHD 患者的症状无特异性,早期可无显著症状。主要症状为呼吸困难、活动耐力下降以及心律失常引起的心悸。晚期可出现咯血、右心衰竭相关症状和猝死。
5.2 PAH-CHD 的体征
PAH 早期原有CHD 体征并不消失,但肺动脉瓣区第二心音(P2)增强。随着PAH 进展,P2逐渐增强乃至亢进,原有CHD 杂音逐渐消失,代之以三尖瓣和肺动脉瓣关闭不全杂音。ES 典型体征为中心性发绀,动脉导管未闭(patent ductus arteriosus,PDA)可出现典型的差异性发绀。
6 PAH-CHD 的辅助检查
6.1 心电图
因缺损类型和肺动脉压高低而表现不同。早期心电图可正常,阻力型PAH 期典型表现为右心室肥厚与劳损,可伴右心房增大。室间隔缺损(ventricular septal defect,VSD)和PDA 患者在动力型PAH 期可表现为左心室肥厚或左、右心室肥厚。
6.2 胸部X 线
肺动脉段凸出,肺血管影加深,透视下可见肺门“舞蹈”征,晚期中心肺动脉粗大而周围肺血管影纤细,同时右心房和右心室增大。对于VSD 和PDA 患者,动力型PAH 期可见左心室增大或者左、右心室均增大。
6.3 超声心动图
6.3.1 超声心动图的作用 (1)发现心脏缺损:随着PAH的进展,CHD 典型体征逐渐消失,超声心动图有助于发现缺损;(2)测量心脏各腔室和大动脉直径,判断PAH 严重程度;(3)测量三尖瓣和肺动脉瓣反流速度,估测肺动脉压力;(4)测量缺损分流大小和方向,评估PAH 严重程度;(5)M 超和多普勒超声尚可测量三尖瓣环收缩期位移和右心室心肌运动指数(Tei 指数),评估患者预后[9];(6)三维超声可测量右心室射血分数,评估右心室功能[10]。
6.3.2 超声心动图的局限性 (1)无法准确测量肺血管压力和阻力;(2)可由于分流消失而漏诊,尤其是PDA[11]。
6.4 计算机断层扫描(CT)
有助于发现各种心脏畸形,判断血管畸形具有独特的优势,但无法检测血流动力学指标。
6.5 心脏磁共振成像(MRI)
可多角度成像,也可评价右心室质量、体积、功能和血流量,有高度的可重复性,目前认为具有应用价值的MRI 标志包括室间隔曲度的改变、右心室射血分数、右心室体积和心脏指数等[12]。MRI 缺点在于耗时长,患者必须长时间保持静止不动,也不能检测血流动力学指标。
6.6 心血管造影
为复杂CHD 诊断的金标准,可确诊心血管畸形及肺动脉发育情况。
6.7 右心导管检查术
为判断患者能否手术治疗和预后的最重要检查方法。常用端孔导管,但Swan-Ganz 导管测量肺楔压(pulmonary wedge pressure,PWP)更有优势。PAH-CHD 患者必须同时测量左、右心系统各腔室压力和血氧含量以及PWP。使用左心房压或左心室舒张末压替代PWP 与实际值存在差距。通常采用Fick 法计算心输出量,然后计算Qp/Qs、肺血管阻力(pulmonary vascular resistance,PVR)、Rp/Rs 等指标。PVR是国际上衡量手术指征的重要指标之一。此外,Pp/Ps 和PVR 指数[PVRI=PVR×体表面积(Wood 单位·m2)]也是主要评估指标。
6.8 急性肺血管反应试验
通过吸入选择性肺血管扩张剂,评价肺血管反应性和病变严重程度,对判断患者预后具有重要作用。建议对重度PAH 同时Qp/Qs≤1.5 的患者实施急性肺血管扩张试验。若PVR 显著升高,即使Qp/Qs >1.5,也可考虑行急性肺血管扩张试验,以判断患者预后。
6.8.1 急性肺血管扩张试验常用试剂 (1)一氧化氮(NO):对体循环无显著影响,2009 年美国心脏病学会基金会(ACCF)/美国心脏协会(AHA)PAH 专家共识[13]将NO 作为急性肺血管扩张试验首选试剂,推荐浓度为10 ~80 ppm(通常20 ~40 ppm,1 ppm=10-6),吸入时间为15 min。但国内没有医用NO,而且操作复杂。(2)腺苷:起始剂量50 μg/(kg·min),每2 ~3 min 剂量增加25 μg/(kg·min),直至患者出现不适或达到最大剂量[200 ~300 μg/(kg·min)][14]。终止试验指征:①肺动脉压下降达到目标值。②体循环收缩压下降30%或<85 mmHg。③用药达到预期的最大剂量。④心率增加40%以上或>100 次/min,或者<60 次/min并出现低血压症状。⑤与用药前相比,右心房压增加20% ~50%,或心脏指数减少>10%。⑥出现不能耐受的不良反应,如恶心、潮红、头痛、胸闷等。腺苷作为急性肺血管扩张剂的缺点是不良反应较多,对PAH-CHD 肺循环和体循环压力均存在显著影响[15]。(3)吸入型伊洛前列素:体重40 kg 以下患者25 ng/(kg·min);体重40 kg 以上患者1 μg/min,加相等体积生理盐水稀释,吸入10 min。终止试验指征:①体循环收缩压降至90 mmHg 以下;②右心房压升高20% ~50%,心脏指数减少>10%;③出现无法耐受的不良反应,如恶心、潮红或头痛。研究显示,吸入型伊洛前列素不良反应较少,能选择性降低PAH-CHD 患者PVR 而对体循环基本无影响,并能改善肺换气功能[14]。
6.9 封堵试验
封堵试验是利用封堵器临时关闭缺损,观察血流动力学变化,从而判断其预后的一种方法。此法多用于孤立性PDA和房间隔缺损(atrial septal defect,ASD)患者,由于受解剖条件限制,VSD 未见有采用该方法的报道。
6.9.1 操作方法 在全心导管的基础上,采用球囊或封堵器暂时关闭缺损,同时监测患者症状、生命体征和血流动力学变化。待完全阻断分流后观察PAP 等血流动力学指标变化,若PAP 无显著下降,或患者出现胸痛、心率和血压下降等变化,说明肺血管病变严重,预后差,不宜关闭缺损;反之,说明PAH 仍主要依赖左向右分流,预后良好,可以彻底关闭缺损。
6.9.2 缺点 存在一定危险性,有诱发PH 危象可能,目前尚未探索出可关闭缺损的确切指标和截点。
7 PAH-CHD 的预后评估
PAH-CHD 的预后评估与其他类型的PAH 相同(表3)。

表3 先天性心脏病相关性肺动脉高压预后评估表
8 PAH-CHD 的治疗
根据PAH 程度决定PAH-CHD 治疗方案:对于动力型PAH 患者,手术关闭缺损是解决PAH 的根本方法;阻力型PAH 可采用靶向药物治疗和心肺联合移植或肺移植联合心脏缺损修补术;对于直接关闭缺损危险性大的“边缘型”PAH患者,可先给予靶向药物治疗或行封堵试验,观察血流动力学变化,然后确定治疗方案。以下治疗主要针对阻力型PAH 而言。
8.1 基础治疗
主要目的是改善右心功能和防治血栓形成,对肺血管病变并无作用。(1)洋地黄类:常用制剂有地高辛和西地兰,可增强心肌收缩力,改善右心功能,并减慢心率。由于患者右心功能差,肝代谢能力降低,建议采用小剂量给药方式。(2)利尿剂:减轻右心负荷,推荐小剂量使用。对于发绀患者,若血红蛋白显著升高,不建议长期使用利尿剂。(3)抗凝药物:主要针对原位血栓,并防止肺动脉血栓形成。常用药物为华法林,建议从小剂量开始使用,逐渐加量,将国际标准化比值(international normalized ratio,INR)维持在1.5 ~2.5。咯血患者忌用。(4)多巴胺和多巴酚丁胺:是治疗重度右心功能衰竭的首选药物,血压偏低首选多巴胺,血压较高首选多巴酚丁胺。两种药物的推荐起始剂量为2 μg/(kg·min),逐渐加量至8 μg/(kg·min)。根据患者具体情况可选择其中一种或联合使用。
8.2 非靶向药物治疗
(1)他汀类:动物实验显示,辛伐他汀能减轻甚至逆转PAH 以及肺血管重构[19-20]。临床研究显示,辛伐他汀可改善PAH 患者心功能,降低肺动脉压[21]。但无临床证据显示PAH-CHD 患者可从他汀类药物治疗中获益。(2)钙通道阻滞剂:研究显示,PAH-CHD 不能从钙通道阻滞剂治疗中获益[16],不推荐使用。
[36]Royal Government of Cambodia,“Political Platform of Royal Government of Cambodia of the Fifth Legislature of National Assembly”, https://www.cambodianembassy.org.uk/f_home/PDF/Political_Platform_Royal_Government_Cambodia_5th.pdf, 2013年9月。
8.3 靶向药物治疗
目前有如下四类,总体选择原则与其他类型PAH 相似,强调依据世界卫生组织(WHO)功能分级进行选择。但PAH-CHD 与其他PAH 存在显著差别。因此,主要讨论PAH-CHD 靶向治疗特点与循证依据。
8.3.1 前列环素类药物 前列环素可抑制血管平滑肌细胞生长和血小板聚集,使血管平滑肌细胞内环腺苷酸(cyclic adenosine monophosphate,cAMP)增加而松弛血管。目前国内仅有吸入型伊洛前列素、贝前列素片和曲前列尼尔注射剂三种制剂,国外还有依前列醇。(1)依前列醇:长期应用依前列醇治疗纽约心功能分级(new york heart function assessment,NYHA)Ⅲ~Ⅳ级的PAH 患者,可改善活动能力、血流动力学及生存率,应用依前列醇治疗特发性PAH 患者,1、2 和3 年生存率分别为87.8%、76.3%和62.8%[22]。雾化吸入依前列醇对治疗PAH 也有效[23],但其半衰期短,需持续吸入,浓度为10 ~50 ng/(kg·min)。虽然无多中心大规模临床对照试验证据,但小样本临床研究显示,依前列醇治疗PAH-CHD 同样效果良好[24-26]。而且,即使婴幼儿应用也非常安全[27]。(2)曲前列尼尔(曲前列环素):是一种长效前列腺Ⅰ-2 类似物,作用时间长达3 h。美国批准其治疗NYHA 心功能Ⅱ~Ⅳ级PAH 患者,欧洲批准其用于NYHA心功能Ⅱ~Ⅲ级PAH 患者。研究显示,皮下注射曲前列尼尔治疗NYHA 心功能Ⅱ~Ⅲ级的特发性肺动脉高压(idiopathic pulmonary arterial hypertension,IPAH)、PAH-CHD和结缔组织病相关性PAH 患者,临床表现均显著改善[28-29]。与依前列醇一样,也可安全应用于婴幼儿。目前美国已批准曲前列尼尔口服制剂用于治疗PAH[30]。(3)伊洛前列素:伊洛前列素化学性质稳定,雾化吸入后可选择性作用于肺血管。推荐剂量为10 ~20 μg/次,6 ~9 次/d。雾化吸入伊洛前列素治疗后,PAH 患者2 年生存率为91%[31],高于历史对照人群的预期生存率63%。治疗PAH-CHD 耐受性良好,且能改善患者生活质量、右心室功能和运动耐量[32-33]。(4)贝前列素:贝前列素口服可改善PAH 患者活动能力与症状,但血流动力学与心功能分级无显著改善,治疗PAH 存在争议。既往认为其疗效随用药时间延长而降低,但最近日本有研究显示,口服大剂量贝前列素治疗特发性和结缔组织病相关性PAH 长期效果良好[34],但无证据显示PAH-CHD 可从中获益。
8.3.2 内皮素受体拮抗剂 内皮素-1(endothelin-1,ET-1)通过ET-A 受体和ET-B 受体起作用。目前,双重受体拮抗剂有波生坦和马西替坦两种,而选择性ET-A 受体拮抗剂仅有安立生坦。(1)波生坦:成人第1 个月62.5 mg/次、每日2 次,若无不良反应,则增至125 mg/次、每日2 次,20 ~40 kg和10 ~20 kg 体重患者的剂量分别为正常体重成人的1/2 和1/4。研究显示,波生坦可改善ES 患者运动能力和血流动力学[35-36]。研究发现,使用4 年之久,患者运动耐力和生活质量仍呈改善状态[37]。波生坦的主要不良反应为影响肝功能,建议使用本药的患者长期监测肝功能。(2)安立生坦:对ET-A 受体拮抗作用较ET-B 受体强4000 倍,可以显著改善PAH 患者血流动力学、生活质量和生存期[38-39],对肝功能无显著影响[40-41]。推荐剂量为5 ~10 mg/次,每日1 次。(3)马西替坦:作为双重受体阻滞剂,在减轻肺纤维化,阻断内皮素受体方面显著强于波生坦,而且对胆盐排出泵无阻断作用。欧美国家已批准可用于PAH 治疗[42],推荐剂量为10 mg/次,每日1 次。
8.3.3 磷酸二酯酶-5(phosphodiesterase-5,PDE-5)抑制剂PDE-5 抑制剂通过抑制环鸟苷酸(cyclic guanosinemonophosphate,cGMP)分解,增加NO 含量而起作用。(1)西地那非:多个研究显示,西地那非治疗PAH-CHD 安全有效[43-44]。其作用特点是疗效随时间延长而降低,需加大剂量方可维持疗效,但随着剂量加大,不良反应也增多[45-46],尤其是17 岁以下患者[47]。因此,目前欧美国家不推荐采用大剂量方式治疗PAH。欧洲药监局批准剂量如下:体重>20 kg患者,20 mg/次,每日3 次,口服;体重≤20 kg 患者,10 mg/次,每日3 次,口服。此外,西地那非也可用于预防停止吸入NO 时的PAP 反弹[48]。(2)伐地那非和他达拉非:在PAH 患者中,伐地那非在初期显示出最大的快速效应,但缺乏像西地那非和他达拉非那样的肺选择性,他达拉非对肺血管的扩张反应最为持久;与西地那非相比,伐地那非和他达拉非并不能提高动脉氧合作用[49-50]。目前,伐地那非尚未批准用于PAH 治疗,也无治疗PAH-CHD 证据。他达拉非已被美国FDA 批准用于PAH 治疗,推荐剂量40 mg/次,每日1 次,口服。但国内多为10 ~20 mg/d。有研究显示,他达拉非治疗PAH-CHD 效果良好[51-52],且治疗儿童患者同样安全有效[53]。注意事项:PDE-5 抑制剂不良反应包括头痛、脸红、消化不良和低血压,与硝酸酯类、抗高血压药同时服用可致严重低血压。
8.3.4 鸟苷酸环化酶激动剂 能够直接刺激鸟苷酸环化酶,增强其对低水平NO 敏感性。目前唯一试剂为利奥西胍(riociguat),对PAH 和血栓栓塞性PH 均有效,已获美国FDA 批准用于PAH 治疗[54-55]。治疗开始剂量为1 mg/次,每日3 次,每间隔不短于2 周的时间增加0.5 mg,直至最大耐受剂量2.5 mg。目前尚无针对利奥西胍治疗PAH-CHD的临床试验。
8.3.5 联合治疗 联合作用机制不同的药物,理论上可以增强治疗效果,但具体如何联合以达到最佳效果,目前尚无明确证据,其安全性和性价比也有待进一步研究。证据比较充分的是,前列环素类与PDE-5 抑制剂联合应用安全、有效[56-57],而其他联合治疗是否有效尚有争议。
8.4 心脏移植、心肺联合移植或肺移植联合心脏缺损修补术
为治疗ES 终极手段,术后免疫排斥反应多见,尚无与靶向药物治疗比较资料。鉴于肺移植5 年存活率仅50%的事实[58],仅推荐终末期ES 且各种靶向药物治疗无效的患者进行此项治疗。
9 ASD 合并PAH
ASD 患者PAH 发生率为16% ~18%,男女比例1∶4,中-重度PAH 约占27%,因PAH 而导致右向左分流者占6% ~13%,出现ES 概率仅为心室水平和肺动脉水平分流型CHD的1/6[59]。预期寿命40 ~50 岁,从出现症状到死亡时间为1 ~19 年,平均时间8 年[60-62]。在20 ~40 岁患者中,有14%出现PAH 后呈现进行性发展,即使关闭缺损,也不能阻止PAH 发展[63-64]。PAH 与缺损类型、位置、大小和年龄等因素相关。儿童期因ASD 而出现PAH 罕见,20 岁以下PAH 发生率仅4%,20 ~40 岁发生率为18%,而40 岁以上发生率高达40.0% ~49.4%。以40 岁为界限,年轻患者的PAH 较年老者更为严重[59]。缺损类型和位置对患者是否发展为PAH存在显著影响[60,65]:原发孔型ASD 患者PAH 发生率可达43% ~57%;继发孔型ASD 患者PAH 发生率差异较大,静脉窦型缺损为26%,中央型ASD 为9%。部分患者PAH 严重程度与ASD 大小无关,表现为PAH 严重而ASD 偏小,目前更倾向将其归为IPAH 之中[66-68]。
9.1 ASD 合并PAH 的介入治疗
(1)ASD 合并高压力低阻力型PAH,选择介入或手术治疗。(2)ASD 伴显著三尖瓣反流、房水平双向分流,若Pp/Ps≤0.8,可考虑试封堵术。完全关闭ASD 后,若PAP 下降25%以上,而主动脉压力无显著下降,动脉血氧饱和度(arterial oxygen saturation,SaO2)升高至94%以上,三尖瓣反流减轻,可考虑永久关闭ASD[69]。(3)ASD 伴左心室腔小,建议使用带孔封堵器实施封堵术。研究显示,采用带孔封堵器不仅可以防止因左心室前负荷突然大量增加而出现急性左心功能不全和心律失常,若术后PAP 升高,残留小孔还能对此进行有效缓冲,防止PAP 急剧升高[70]。对于巨大ASD采用带孔封堵器封堵术后随访PAP 恢复正常者,可考虑再使用封堵器将遗留的小孔关闭。
9.2 疗效评价
ASD 封堵术后早期症状改善并不代表患者长期预后良好[71]。虽然中-重度PAH 患者实施ASD 封堵术后短期内能够存活,PAP 也呈降低趋势,但仅43.6% 患者可降至40 mmHg 以下,15.4%患者呈现持续性重度PAH[69]。
10 VSD 合并PAH
VSD 患者PAH 发生率报道不一致[2,72]。成年VSD 患者PAH 总体发生率达39%,出现PAH 但未达到ES 标准者为11%,ES 为28%。10 岁以前ES 发生率约10%。是否引起PAH 取决于缺损大小和肺血管床状态,而非缺损位置。成年VSD 合并ES 的患者往往在30 多岁死亡。已行VSD 修补术的患者PAH 总体发生率约2%,成年患者术后PAH 发生率约13%。
10.1 VSD 合并PAH 的介入/手术治疗方法
由于婴幼儿外科手术风险较高,而且,即使是非限制型VSD,1 周岁以内也很少发生阻力型PAH[73]。因此,对于婴幼儿VSD 相关性PAH 而言,若心力衰竭不严重,应持续药物治疗6 个月再确定治疗方案,若PAP 未恢复正常范围,或虽恢复正常但患儿仍有症状,应考虑手术治疗。2 岁以上儿童凡Pp/Ps >0.5、mPAP >25 mmHg,均应行介入或手术治疗[74]。2010 年ESC 成人CHD 管理指南[6]VSD 合并PAH 手术治疗适应证选择见表4。

表4 VSD 合并PAH 手术治疗适应证
10.2 疗效评价[74-78]
VSD 合并PAH 手术效果与年龄有重要关系,2 岁以下患儿术后常可恢复正常,2 岁以上合并重度PAH 远期疗效不肯定。在补片上打孔,或采用带有阀门式的补片修补,效果并不确定。VSD 相关性ES 患者心肺联合移植术后1 年、5 年和10 年生存率分别是61%、40% 和25%,优于其他CHD 和IPAH。
11 PDA 合并PAH
PDA 患者PAH 发病率显著高于VSD 和ASD,若不实施关闭术,除细小型和哑型PDA 外,几乎均可导致PAP 升高,ES 发生率可达50%。
11.1 PDA 合并重度PAH 的介入治疗
对于合并重度PAH 患者,可采用封堵试验[79]判断肺血管病变程度及手术后PAH 变化。封堵试验对象为Qp/Qs >1.5 且股动脉SaO2>90%者。多数学者以试封堵后肺动脉收缩压(systolic pulmonary arterial pressure,sPAP)下降超过30%为封堵术指标,也有学者认为,若封堵后sPAP 或mPAP降低20% 或30 mmHg 以上,PVR 下降,而主动脉压力和SaO2无下降或上升,且无全身反应,可进行永久封堵。新近研究显示,以试封堵术后肺/体循环收缩压比值作为判断指标最为可靠,如试封堵术后肺/体循环收缩压比值<0.5,可永久关闭PDA,术后PAP 最终将完全恢复正常;反之,如比值>0.5,即使封堵术后PAP 显著下降,也必然存在术后持续性PAH[80]。
11.2 疗效评价
短期效果令人满意,但无PDA 合并重度PAH 介入治疗的多中心、大规模、长期随访研究结果。
12 复杂先天性心脏病(complex congenital heartdisease,CCHD)相关性PAH
CCHD 约占CHD 的30%。PAH 一般见于肺血增多型CCHD。由于多合并较大或多水平体-肺分流,PAH 发病年龄早,往往一出生时就存在严重PAH。PAH 程度除与分流水平、体-肺分流量、分流持续时间等因素相关外,也与病变血流动力学特点密切相关[81-83]。对于CCHD 患者,无论是否接受外科手术治疗,PAH 存在与否对患者预后起决定性作用。
12.1 治疗
关键在于适当时机进行手术治疗。对于合并显著分流的CCHD,一旦发现,应在体重允许条件下尽早手术,防止发生严重PAH,最适宜手术年龄为出生后1 ~3 个月。肺动脉环缩术(Banding 术)是常用姑息手术方法,适用于新生儿、婴幼儿肺血增加有PAH 趋势或有显著PAH,但目前条件不能根治或不适合立即根治者。
12.2 预后
预后较简单分流性CHD 差,若不早期干预,仅有少数患者能存活至成年。预后与心脏畸形复杂程度和治疗时机密切相关,其中完全性房室间隔缺损、主肺间隔缺损和主动脉弓离断预后较好。
13 围术期相关性PH
手术创伤和体外循环(cardiopulmonary bypass,CPB)会诱发全身炎性反应,导致肺血管内皮细胞受损,血栓素A2、ET-1 等缩血管的细胞因子增多,PVR 增高,诱发PAH[84]。合并PAH 的CHD 患者术后ET-1 下降至正常水平需要48 h左右[84-85]。术后早期(<30 d)PAP 高于正常,称为术后反应性肺高压(reactive pulmonary hypertension,RPH)[86]。术后RPH 以及肺高压危象(pulmonary hypertension crisis,PHC)是CHD 术后早期常见并发症及死亡原因[87-88]。致命性PH 及PH 危象发生率分别为2.0%和0.7%,但在完全性房室间隔缺损伴Down's 综合征患者其发生率仍偏高。
13.1 诊断
13.1.1 诊断标准 (1)术后RPH:海平面安静状态时mPAP≥25 mmHg。(2)PHC:PAP 急剧升高,超过体动脉压力,伴或不伴有体动脉压力下降;CO 和SaO2显著下降。
13.1.2 术前高危因素 (1)年龄(按病种分类):>6 个月(完全性大血管转位/全肺静脉异位引流/永存动脉干/主动脉弓离断/单心室),>1 岁(完全性房室间隔缺损/右心室双出口),>2 岁(VSD/PDA),>4 岁(ASD)。(2)临床表现:呼吸道感染减少,伴心功能(活动量)降低;渐进性青紫,SaO2<95%(不吸氧时)。(3)辅助检查:胸部X 线示,心影缩小,肺动脉段突出。心电图示,电轴右偏,右心室肥厚。超声心动图示,心室、心房、大血管水平双向分流,以右向左分流为主。肺小动脉造影示,肺动脉分支管径细且不齐,末梢蜷曲,毛细血管充盈差。心导管检查示,Pp/Ps >0.75,Qp/Qs<1.5,PVR >9 woods,PWP <12 mmHg[89]。
13.2 治疗
13.2.1 预防方法 术后RPH 和PHC 重在预防,预防方法见表5。
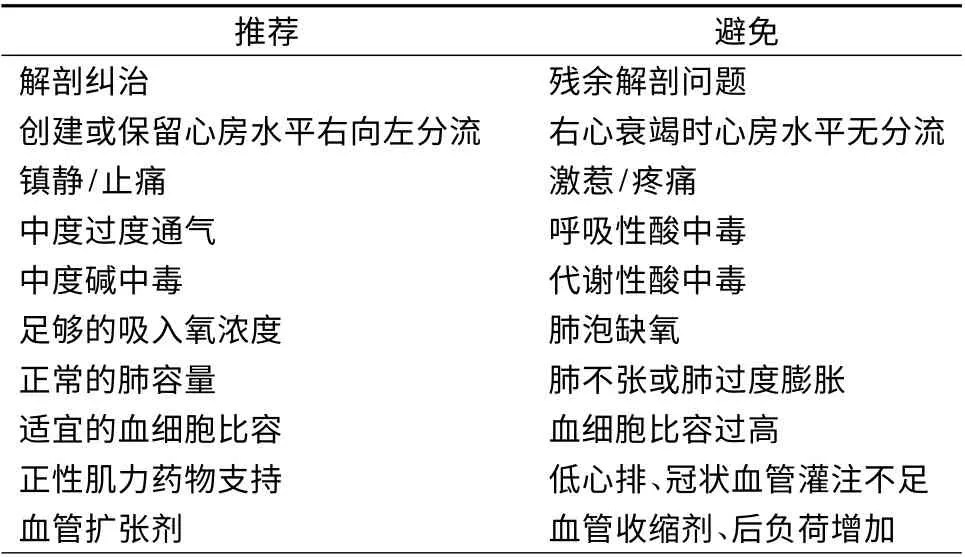
表5 反应性肺高压和肺高压危象预防策略
13.2.2 肺血管扩张剂 对于体-肺分流相关性PAH,在常规支持治疗的基础上,如何针对不同发病环节使用特异性肺血管扩张剂证据较少[90-94],治疗更多取决于专家经验[95]。因负性肌力作用和体循环阻力下降可加重病情,儿科病例不推荐使用钙通道阻滞剂。存在肺静脉回流梗阻或左心室功能不全患儿,禁用NO、伊洛前列素等急性肺血管扩张剂。对于主动脉缩窄、主动脉弓中断合并VSD 者,由于左心系统发育不良可能性小,术后仍可应用伊洛前列素、NO 等急性肺血管扩张剂。
14 术后迟发性PAH
迟发性PAH 是相对于术后RPH 而言,这类患者因术前即存在重度PAH,术后PAP 并未完全降至正常,经过一段时间后PAH 再次加重而出现右心衰竭症状。若术后PAP 降至正常,数年后再次出现PAH,则更倾向于IPAH。由于术前PAP 显著升高,即使分流中断后,PAH 导致的肺血管床恶性重构也需要较长一段时间方可逐渐恢复。此外,部分患者术前还可能存在左心功能不全(如大型PDA),或阻断分流后因负荷增加而出现左心功能不全(如大型ASD)。在这些因素作用下,即使患者为动力型PAH,术后PAP 也需要相当长一段时间方可恢复正常。因此,并非所有术后PAP 升高者均为术后PAH。对于术后迟发PAH 如何界定,目前尚未明确,以术后6 个月mPAP 仍然高于25 mmHg 作为术后迟发PAH 时间界限较为合适。注册研究显示,VSD 关闭术后PAH 发生率约2%,继发孔型ASD 术后发生率约3%[2,82,96]。根据手术方式,迟发性PAH 可分为两种:(1)完全矫正术后PAH,指CHD 外科矫治术后PAH,可在术后即刻、数月或数年后出现,同时没有残余分流或与外科手术相关的后遗症。(2)不完全矫正术后PAH,包括功能性单心室行双向腔肺分流术(bidirectional cavopulmonary shunt,BCPS)和全腔静脉-肺动脉连接术(total cavopulmonary connection,TCPC)术后。
14.1 致病因素
肺血管床功能和结构状况是决定患者症状和预后的关键因素。术后迟发性PAH 可能与手术时机过迟、误判手术可能性、右心室后负荷长期作用导致结构重塑不可逆转,以及肺血管发育情况等有关,有以下两种情形:(1)术前肺血增多。由于大量左向右分流造成肺血管床阻力增高,出现肺血管收缩和重构。虽然术后心内体-肺分流被阻断后,PAP 可以暂时下降,但个别患者其丛样血管病变无法逆转,甚至在仍然偏高的PAP 作用下还会再次进展,导致PAH 恶化。(2)术前肺血少的发绀型CHD。由于长期缺乏正常的肺血流灌注,其肺血管床往往发育不良。如肺动脉闭锁患者,虽然其左右肺动脉的发育可以达到根治术标准,但往往存在肺内叶、段动脉缺失,根治术后这种发育欠佳的肺血管床难以容纳突然增加的肺血流而使PAP 持续偏高,导致节段性PAH 形成。
14.2 预后
预后很差,预期寿命甚至短于未手术的CCHD 和ES 患者,较ES 患者累积生存时间缩短1.3 年。
15 结语
PAH-CHD 作为PAH 的一种,却与其他类型PAH 存在显著差异,主要表现在以下几个方面:(1)年龄差异较大,其他类型PAH 多以成人为主,而PAH-CHD 随年龄增长、畸形矫治时间的拖延而逐渐加重[97];(2)发病机制和血流动力学变化独特,系体-肺分流导致肺血管内皮受损所致,发病的早晚,进展的快慢以及预后的好坏与缺损的位置、大小以及病变的复杂程度等多种因素相关[98];(3)不同PAH 阶段,治疗方法不同,早期为动力型PAH,关闭缺损是关键,而一旦成为阻力型PAH,关闭缺损反而有害;(4)由于血流动力学与其他类型PAH 显著不同,对PAH 靶向药物的反应也不同,对其他PAH 反应良好的制剂未必可以直接转用于PAHCHD 上。近年来,随着各种PAH 靶向药物的问世,ES 患者预后显著改善[99-100]。但目前大部分PAH 靶向药物研究均以成人和其他类型PAH 为对象,有关治疗PAH-CHD 的循证数据相对缺乏,尤其是儿童PAH-CHD方面,数据非常有限,今后应在儿童PAH-CHD 方面加强多中心、大规模临床研究,为靶向药物治疗儿童PAH-CHD 的效果和安全性提供更多依据。
写作组成员(按姓氏拼音排序):高伟(上海儿童医学中心),顾红(首都医科大学附属北京安贞医院),胡大一(北京大学人民医院),华益民(四川大学华西第二医院),黄奕高(广东省人民医院),李奋(上海儿童医学中心),秦永文(第二军医大学附属长海医院),宋治远(第三军医大学西南医院),王广义(中国人民解放军总医院),吴炳祥(哈尔滨医科大学附属第二医院),徐仲英(中国医学科学院阜外心血管病医院),徐卓明(上海儿童医学中心),张端珍(中国人民解放军沈阳军区总医院),张伟华(昆明市延安医院),张玉顺(西安交通大学第一附属医院),周达新(复旦大学附属中山医院),朱鲜阳(中国人民解放军沈阳军区总医院)
专家组成员(按姓氏拼音排序):冯元(四川大学华西医院),高伟(上海儿童医学中心),顾红(首都医科大学附属北京安贞医院),管丽华(复旦大学附属中山医院),韩雅玲(中国人民解放军沈阳军区总医院),胡大一(北京大学人民医院),华益民(四川大学华西第二医院),黄奕高(广东省人民医院),荆志成(中国医学科学院阜外心血管病医院),李奋(上海儿童医学中心),刘迎龙(首都医科大学附属北京安贞医院),马依彤(新疆医科大学第一附属医院),潘欣(上海胸科医院),秦永文(第二军医大学附属长海医院),宋治远(第三军医大学西南医院),苏俊武(首都医科大学附属北京安贞医院),吴炳祥(哈尔滨医科大学附属第二医院),伍广伟(广西壮族自治区人民医院),伍伟峰(广西医科大学第一附属医院),王广义(中国人民解放军总医院),王慧深(中山大学附属第一医院),徐仲英(中国医学科学院阜外心血管病医院),徐卓明(上海儿童医学中心),杨天和(贵州省人民医院),余再新(中南大学湘雅医院),曾智(四川大学华西医院),张曹进(广东省人民医院),张端珍(中国人民解放军沈阳军区总医院),张刚成(武汉亚洲心脏病医院),张伟华(昆明市延安医院),张玉顺(西安交通大学第一附属医院),张智伟(广东省人民医院),赵世华(中国医学科学院阜外心血管病医院),赵仙先(第二军医大学附属长海医院),周达新(复旦大学附属中山医院),朱鲜阳(中国人民解放军沈阳军区总医院)
[1]Galie N,Manes A, Palazzini M, et al. Management of pulmonary arterial hypertension associated with congenital systemic-to-pulmonary shunts and Eisenmenger' s syndrome.Drugs,2008,68:1049-1066.
[2]Duffels MG,Engelfriet PM,Berger RM,et al. Pulmonary arterial hypertension in congenital heart disease: an epidemiologic perspective from a Dutch registry. Int J Cardiol,2007,120:198-204.
[3]Diller GP,Gatzoulis MA. Pulmonary vascular disease in adults with congenital heart disease. Circulation,2007,115:1039-1050.
[4]兰贝蒂,张玉顺. 肺动脉高压合并小缺损先天性心脏病的认识与研究进展.心血管病学进展,2010,31:751-753.
[5]丁仲如,秦永文. 先天性心脏病合并肺动脉高压分级及性质的评估.介入放射学杂志,2008,17:523-526.
[6]Baumgartner H,Bonhoeffer P,De Groot NM,et al. ESC Guidelines for the management of grown-up congenital heart disease (new version 2010). Eur Heart J,2010,31:2915-2957.
[7]Diller GP,Dimopoulos K,Kafka H,et al. Model of chronic adaptation:right ventricular function in Eisenmenger syndrome.Eur Heart J,2007,9:54-60.
[8]Daliento L,Somerville J,Presbitero P,et al. Eisenmenger syndrome. Factors relating to deterioration and death. Eur Heart J,1998,19:1845-1855.
[9]Forfia PR,Fisher MR,Mathai SC,et al. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am J Respir Crit Care Med,2006,174:1034-1041.
[10]Inaba T,Yao A,Nakao T,et al. Volumetric and functional assessment of ventricles in pulmonary hypertension on 3-dimensional echocardiography. Circ J,2012,77:198-206.
[11]Krichenko A,Benson LN,Burrows P,et al. Angiographic classification of the isolated,persistently patent ductus arteriosus and implications for percutaneous catheter occlusion. Am J Cardiol,1989,63:877-880.
[12]Van Wolferen SA,Marcus JT,Boonstra A,et al. Prognostic value of right ventricular mass,volume,and function in idiopathic pulmonary arterial hypertension. Eur Heart J,2007,28:1250-1257.
[13]McLaughlin VV,Archer SL,Badesch DB,et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension:a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association:developed in collaboration with the American College of Chest Physicians,American Thoracic Society,Inc.,and the Pulmonary Hypertension Association. Circulation,2009,119:2250-2294.
[14]Zhang DZ,Zhu XY,Meng J,et al. Acute hemodynamic responses to adenosine and iloprost in patients with congenital heart defects and severe pulmonary arterial hypertension. Int J Cardiol,2011,147:433-437.
[15]张端珍,朱鲜阳,崔春生,等. 先天性心脏病重度肺动脉高压对腺苷的急性反应. 心脏杂志,2011,23:241-244.
[16]Montani D,Savale L,Natali D,et al. Long-term response to calcium-channel blockers in non-idiopathic pulmonary arterial hypertension. Eur Heart J,2010,31:1898-1907.
[17]Limsuwan A,Khosithseth A,Wanichkul S,et al. Aerosolized iloprost for pulmonary vasoreactivity testing in children with long-standing pulmonary hypertension related to congenital heart disease. Catheter Cardiovasc Interv,2009,73:98-104.
[18]Lopes AA,O'Leary PW. Measurement,interpretation and use of haemodynamic parameters in pulmonary hypertension associated with congenital cardiac disease. Cardiol Young,2009,19:431-435.
[19]Nishimura T,Faul JL,Berry GJ,et al. Simvastatin attenuates smooth muscle neointimal proliferation and pulmonary hypertension in rats. Am J Respir Crit Care Med,2002,166:1403-1408.
[20]Girgis RE,Li D,Zhan X,et al. Attenuation of chronic hypoxic pulmonary hypertension by simvastatin. Am J Physiol Heart Circ Physiol,2003,285:938-945.
[21]Kao PN. Simvastatin treatment of pulmonary hypertension:an observational case series. Chest,2005,127:1446-1452.
[22]McLaughlin VV,Shillington A,Rich S. Survival in primary pulmonary hypertension:the impact of epoprostenol therapy.Circulation,2002,106:1477-1482.
[23]Buckley MS,Feldman JP. Inhaled epoprostenol for the treatment of pulmonary arterial hypertension in critically ill adults.Pharmacotherapy,2010,30:728-740.
[24]Rosenzweig EB,Kerstein D,Barst RJ. Long-term prostacyclin for pulmonary hypertension with associated congenital heart defects. Circulation,1999,99:1858-1865.
[25]Fernandes SM,Newburger JW,Lang P,et al. Usefulness of epoprostenol therapy in the severely ill adolescent/adult with Eisenmenger physiology. Am J Cardiol,2003,91:632-635.
[26]Thomas IC,Glassner-Kolmin C,Gomberg-Maitland M. Longterm effects of continuous prostacyclin therapy in adults with pulmonary hypertension associated with congenital heart disease. Int J Cardiol,2013,168:4117-4121.
[27]McIntyre CM,Hanna BD, Rintoul N,et al. Safety of epoprostenol and treprostinil in children less than 12 months of age. Pulm Circ,2013,3:862-869.
[28]Benza RL,Seeger W,McLaughlin VV,et al. Long-term effects of inhaled treprostinil in patients with pulmonary arterial hypertension:the Treprostinil Sodium Inhalation Used in the Management of Pulmonary Arterial Hypertension (TRIUMPH)study open-label extension. J Heart Lung Transplant,2011,30:1327-1333.
[29]Sadushi-Kolii R,Skoro-Sajer N,Zimmer D,et al. Long-term treatment, tolerability, and survival with sub-cutaneous treprostinil for severe pulmonary hypertension. J Heart Lung Transplant,2012,31:735-743.
[30]Hill NS,Badesch D,Benza RL,et al. Perspectives on oral pulmonary hypertension therapies recently approved by the food and drug administration. Ann Am Thorac Soc,2015.[Epub ahead of print]
[31]Olschewski H,Hoeper MM,Behr J,et al. Long-term therapy with inhaled iloprost in patients with pulmonary hypertension.Respir Med,2010,104:731-740.
[32]Yang SI,Chung WJ,Jung SH,et al. Effects of inhaled iloprost on congenital heart disease with Eisenmenger syndrome. Pediatr Cardiol,2012,33:744-748.
[33]Cha KS,Cho KI,Seo JS,et al. Effects of inhaled iloprost on exercise capacity,quality of life,and cardiac function in patients with pulmonary arterial hypertension secondary to congenital heart disease (the Eisenmenger syndrome)(from the EIGER Study). Am J Cardiol,2013,112:1834-1839.
[34]Sakao S,Tanabe N,Kasahara Y,et al. Long-term survival of Japanese patients with pulmonary arterial hypertension treated with beraprost sodium,an oral prostacyclin analogue. Intern Med,2014,53:1913-1920.
[35]Galiè N,Beghetti M,Gatzoulis MA,et al. Bosentan therapy in patients with Eisenmenger syndrome:a multicenter,doubleblind, randomized, placebo-controlled study. Circulation,2006,114:48-54.
[36]Berger RM,Beghetti M,Galiè N,et al. Atrial septal defects versus ventricular septal defects in BREATHE-5,a placebocontrolled study of pulmonary arterial hypertension related to Eisenmenger's syndrome:a subgroup analysis. Int J Cardiol,2010,144:373-378.
[37]Vis JC,Duffels MG,Mulder P,et al. Prolonged beneficial effect of bosentan treatment and 4-year survival rates in adult patients with pulmonary arterial hypertension associated with congenital heart disease. Int J Cardiol,2013,164:64-69.
[38]Badesch DB,Feldman J, Keogh A, et al. ARIES-3:ambrisentan therapy in a diverse population of patients with pulmonary hypertension. Cardiovasc Ther,2012,30:93-99.
[39]Zuckerman WA,Leaderer D,Rowan CA,et al. Ambrisentan for pulmonary arterial hypertension due to congenital heart disease. Am J Cardiol,2011,107:1381-1385.
[40]McGoon MD,Frost AE,Oudiz RJ,et al. Ambrisentan therapy in patients with pulmonary arterial hypertension who discontinued bosentan or sitaxsentan due to liver function test abnormalities. Chest,2009,135:122-129.
[41]Condliffe R,Elliot CA,Hurdman J,et al. Ambrisentan therapy in pulmonary hypertension:clinical use and tolerability in a referral centre. Ther Adv Respir Dis,2014,8:71-77.
[42]Patel T,McKeage K. Macitentan:first global approval. Drugs,2014,74:127-133.
[43]张端珍,朱鲜阳,贾丽娟,等. 西地那非治疗先天性心脏病相关性肺动脉高压的有效性与安全性. 心脏杂志,2012,24:600-603.
[44]夏燕亮,马如海,陈宏. 西地那非治疗高原地区先天性心脏病合并重度肺动脉高压的疗效和安全性. 中国循环杂志,2014,29:702-705.
[45]Galiè N,Ghofrani HA,Torbicki A,et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med,2005,353:2148-2157.
[46]Rubin LJ,Badesch DB,Fleming TR,et al.Long-term treatment with sildenafil citrate in pulmonary arterial hypertension:the SUPER-2 study. Chest,2011,140:1274-1283.
[47]Maxey DM,Ivy DD,Ogawa MT,et al. Food and Drug Administration (FDA)postmarket reported side effects and adverse events associated with pulmonary hypertension therapy in pediatric patients. Pediatr Cardiol,2013,34:1628-1636.
[48]Matamis D,Pampori S,Papathanasiou A,et al.Inhaled NO and sildenafil combination in cardiac surgery patients with out-ofproportion pulmonary hypertension: acute effects on postoperative gas exchange and hemodynamics. Circ Heart Fail,2012,5:47-53.
[49]Arif SA,Poon H. Tadalafil:a long-acting phosphodiesterase-5 inhibitor for the treatment of pulmonary arterial hypertension.Clin Ther,2011,33:993-1004.
[50]Buckley MS,Staib RL,Wicks LM,et al. Phosphodiesterase-5 inhibitors in management of pulmonary hypertension:safety,tolerability,and efficacy. Drug Healthc Patient Saf,2010,2:151-161.
[51]Bharani A,Patel A,Saraf J,et al. Efficacy and safety of PDE-5 inhibitor tadalafil in pulmonary arterial hypertension. Indian Heart J,2007,59:323-328.
[52]Mukhopadhyay S,Nathani S,Yusuf J,et al. Clinical efficacy of phosphodiesterase-5 inhibitor tadalafil in Eisenmenger syndrome--a randomized, placebo-controlled, double-blind crossover study. Congenit Heart Dis,2011,6:424-431.
[53]Shiva A,Shiran M,Rafati M,et al. Oral tadalafil in children with pulmonary arterial hypertension. Drug Res (Stuttg),2015.[Epub ahead of print].
[54]Ghofrani HA,Galiè N,Grimminger F,et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med,2013,369:330-340.
[55]Conole D,Scott LJ. Riociguat:first global approval. Drugs,2013,73:1967-1975.
[56]Simonneau G,Rubin LJ,Galiè N,et al. Long-term sildenafil added to intravenous epoprostenol in patients with pulmonary arterial hypertension. J Heart Lung Transplant,2014,33:689-697.
[57]张曹进,黄奕高,黄涛,等. 伊诺前列素联合小剂量他达那非治疗成人先天性心脏病并重度肺动脉高压的单中心、开放、对照研究. 中华心血管病杂志,2014,42:474-480.
[58]Yusen RD,Christie JD,Edwards LB,et al. The Registry of the International Society for Heart and Lung Transplantation:Thirtieth Adult Lung and Heart-Lung Transplant Report—2013;focus theme:age. J Heart Lung Transplant,2013,32:965-978.
[59]Besterman E.Atrial septal defect with pulmonary hypertension.Br Heart J,1961,23:587-598.
[60]Vogel M,Berger F,Kramer A,et al. Incidence of secondary pulmonary hypertension in adults with atrial septal or sinus venosus defects. Heart,1999,82:30-33.
[61]Craig RJ,Selzer A. Natural history and prognosis of atrial septal defect. Circulation,1968,37,805-815.
[62]Campbell M.Natural history of atrial septal defect. Br Heart J,1970,32:820-826.
[63]Attie F,Rosas M,Granados N,et al. Surgical treatment for secundum atrial septal defects in patients >40 years old. A randomized clinical trial. J Am Coll Cardiol,2001,38:2035-2042.
[64]O'Donnell C,Ruygrok PN,Whyte K, et al. Progressive pulmonary hypertension post atrial septal defect device closureearly symptomatic improvement may not predict outcome.Heart,Lung and Circulation,2010,19:713-716.
[65]Sachweh JS,Daebritz SH,Hermanns B,et al. Hypertensive pulmonary vascular disease in adults with secundum or sinus venosus atrial septal defect. Ann Thorac Surg,2006,81:207-213.
[66]Galiè N,Hoeper MM,Humbert M,et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J,2009,34:1219-1263.
[67]Roberts KE,McElroy JJ,Wong WP,et al. BMPR2 mutations in pulmonary arterial hypertension with congenital heart disease.Eur Respir J,2004,24:371-374.
[68]Therrien J,Rambihar S,Newman B,et al. Eisenmenger syndrome and atrial septal defect:nature or nurture?Can J Cardiol,2006,22:1133-1136.
[69]Snchez-Recalde ,Oliver JM,Galeote G,et al. Atrial septal defect with severe pulmonary hypertension in elderly patients:usefulness of transient balloon occlusion. Rev Esp Cardiol,2010,63:860-864.
[70]陈火元,朱鲜阳,韩秀敏,等.成人房间隔缺损合并肺动脉高压患者导管封堵术后左心功能变化. 介入放射学杂志,2013,22:365-368.
[71]Providência R,Botelho A, Quintal N, et al. Pulmonary hypertension in patients with ostium secundum atrial septal defect--is it related to echocardiographic complexity?Rev Port Cardiol,2009,28:1087-1096.
[72]Engelfriet PM,Duffels MG,Mller T,et al. Pulmonary arterial hypertension in adults born with a heart septal defect:the Euro Heart Survey on adult congenital heart disease. Heart,2007,93:682-687.
[73]Rosenzweig EB,Barst RJ. Congenital heart disease and pulmonary hypertension:pharmacology and feasibility of late surgery. Prog Cardiovasc Dis,2012,55:128-33.
[74]Provencher S,Sitbon O,Humbert M,et al. Long-term outcome with first-line bosentan therapy in idiopathic pulmonary arterial hypertension.Eur Heart J,2006,27:589-595.
[75]Serino G,Giacomazzi F,et al. Pulmonary arterial hypertension in adult patients with congenital heart disease. Pediatr Med Chir,2010,32:274-279.
[76]Farber HW,Foreman AJ,Miller DP,et al. REVEAL Registry:correlation of right heart catheterization and echocardiography in patients with pulmonary arterial hypertension. Congest Heart Fail,2011,17:56-64.
[77]Roos-HesselinkJW,Meijbboom FJ,Spitaels SE,et al. Outcome of patients after surgical closure of ventricular septal defect at young age:longitudinal follow-up of 22-34 years. Eur Heart J,2004,25:1057-1062.
[78]Mendelof EN,Meyers BF, Sundt TM, et al. Lung transplantation for pulmonary vascular disease. Ann Thorac Surg,2002,73:209-2l7.
[79]Yan C,Zhao S,Jiang S,et al. Transcatheter closure of patent ductus arteriosus with severe pulmonary arterial hypertension in adults. Heart,2007,93:514-518.
[80]Zhang DZ,Zhu XY,Lv B,et al. Trial occlusion to assess the risk of persistent pulmonary arterial hypertension after closure of a large patent ductus arteriosus in adolescents and adults with elevated pulmonary artery pressure. Circ Cardiovasc Interv,2014,7:473-481.
[81]Friedman WF.Proceedings of National Heart,Lung,and Blood Institute pediatric cardiology workshop: pulmonary hypertension. Pediatr Res,1986,20:811-824.
[82]Beghetti M. Congenital heart disease and pulmonary hypertension. Rev Port Cardiol,2004,23:273-281.
[83]Granton JT,Rabinovitch M. Pulmonary arterial hypertension in congenital heart disease. Cardiol Clin,2002,20:441-457.
[84]Seghaye MC. The clinical implications of the systemic inflammatory reaction related to cardiac operations in children.Cardiol Young,2003,13:228-239.
[85]Kageyama K,Hashimoto S,Nakajima Y,et al. The change of plasma endothelin-1 levels before and after surgery with or without Down syndrome. Paediatr Anaesth,2007,17:1071-1077.
[86]Hopkins RA,Bull C, Haworth SG, et al. Pulmonary hypertensive crises following surgery for congenital heart defects in young children. Eur J Cardiothorac Surg,1991,5:628-634.
[87]Atz AM,Lefler AK,Fairbrother DL,et al. Sildenafil augments the effect of inhaled nitric oxide for postoperative pulmonary hypertensive crises. J Thorac Cardiovasc Surg,2002,124:628-629.
[88]Polderman FN,Cohen J,Blom NA,et al. Sudden unexpected death in children with a previously diagnosed cardiovascular disorder. Int J Cardiol,2004,95:171-176.
[89]Galiè N,Hoeper MM,Humbert M,et al. Guidelines for the diagnosis and treatment of pulmonary hypertension:the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC)and the European Respiratory Society (ERS),endorsed by the International Society of Heart and Lung Transplantation(ISHLT). Eur Heart J,2009,30:2493-2537.
[90]Simonneau G,Barst RJ, Galie N, et al. Continuous subcutaneous infusion of treprostinil,a prostacyclin analogue,in patients with pulmonary arterial hypertension. a doubleblind,randomized,placebo-controlled trial. Am J Respir Crit Care Med,2002,165:800-804.
[91]Rubin LJ,Badesch DB,Barst RJ,et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med,2002,346:896-903.
[92]Galie N,Humbert M,Vachiery JL,et al. Effects of beraprost sodium,an oral prostacyclin analogue, in patients with pulmonary arterial hypertension:a randomized,double-blind placebo-controlled trial. J Am Coll Cardiol,2002,39:1496-1502.
[93]Barst RJ,Langleben D,Badesch D,et al. Treatment of pulmonary arterial hypertension with the selective endothelin-A receptor antagonist sitaxsentan. J Am Coll Cardiol,2006,47:2049-2056.
[94]Barst RJ,Langleben D,Frost A,et al. Sitaxsentan therapy for pulmonary arterial hypertension. Am J Respir Crit Care Med,2004,169:441-447.
[95]Beghetti M,Galiè N. Eisenmenger syndrome: a clinical perspective in a new therapeutic era of pulmonary arterial hypertension. J Am Coll Cardiol,2009,53:733-740.
[96]Bando K,Turrentine MW, Sharp TG, et al. Pulmonary hypertension after operations for congenital heart disease:analysis of risk factors and management. J Thorac Cardiovasc Surg,1996,112:1600-1607.
[97]陈果,何建国,柳志红,等.不同类型肺动脉高压患者临床特征和血流动力学的比较分析.中国循环杂志,2013,28:300-303.
[98]孙云娟,逢坤静,曾伟杰,等. 先天性心脏病相关性肺动脉高压的临床筛查.中华医学杂志,2012,92:1091-1094.
[99]Dimopoulos K,Inuzuka R,Goletto S,et al. Improved survival among patients with Eisenmenger syndrome receiving advanced therapy for pulmonary arterial hypertension. Circulation,2010,121:20-25.
[100]Manes A,Palazzini M,Leci E,et al. Current era survival of patients with pulmonary arterial hypertension associated with congenital heart disease: a comparison between clinical subgroups. Eur Heart J,2014,35:716-724.
猜你喜欢
预防青少年犯罪研究(2022年1期)2022-08-15 00:35:32
心肺血管病杂志(2020年5期)2021-01-14 00:43:38
电子技术与软件工程(2019年21期)2020-01-16 05:55:44
家庭医学(下半月)(2019年10期)2019-11-16 08:59:52
西江月(2018年5期)2018-06-08 05:47:42
电信科学(2017年6期)2017-07-01 15:44:53
海峡姐妹(2017年5期)2017-06-05 08:53:17
中国体外循环杂志(2015年3期)2015-12-08 05:13:00
中国水利(2015年14期)2015-02-28 15:14:16
肝胆胰外科杂志(2015年1期)2015-02-27 11:11:30
