
碱性Li3PO4催化剂的失活及再生
2015-07-07 15:48马卫华
石油化工 2015年4期
李 浩,李 菲,周 璇,马卫华,钟 秦
(南京理工大学 化工学院,江苏 南京 210094)
碱性Li3PO4催化剂的失活及再生
李 浩,李 菲,周 璇,马卫华,钟 秦
(南京理工大学 化工学院,江苏 南京 210094)
研究了气相环氧丙烷(PO)异构化制烯丙醇反应中碱性Li3PO4催化剂的失活及再生。实验结果表明,在280 ℃下反应54 h后,因催化剂表面覆盖大量含氢碳物种引起失活,采用煅烧法、水蒸气洗涤法去除催化剂上的积碳,可使催化剂得到再生。通过XRD、FTIR、原位DRIFT、BET、TG、MAS NMR等手段对催化剂进行表征发现,失活Li3PO4催化剂的比表面及孔体积均下降,但催化剂晶型未发生改变,也无新物相生成。积碳分解温度为300~500 ℃,在350 ℃下积碳分解所需时间为40~50 min。煅烧法再生的Li3PO4催化剂上PO转化率为90.7%,烯丙醇选择性为89.8%,与新鲜Li3PO4催化剂的活性相近。
环氧丙烷; 异构化; 烯丙醇;磷酸锂催化剂; 积碳; 失活; 再生
Li3PO4是一种斜方晶系的白色结晶,有γ型和β型两种特征晶型[1],其中,β-Li3PO4比γ-Li3PO4更稳定[2],但当温度在400~600 ℃内时,β-Li3PO4将会不可逆的转变成γ-Li3PO4[3]。Li3PO4除用作生产彩色荧光粉的理想原料外,还广泛应用于锂电池、光学传感器、特种玻璃和催化剂等方面[4-12]。作为催化剂,Li3PO4主要用于环氧烷烃的异构化反应,具有较高的转化率和选择性。
以Li3PO4为催化剂进行环氧丙烷(PO)异构化反应制烯丙醇(AA)有气相法[13]和液相法[14]两种生产工艺,液相法工艺中催化剂可连续再生,但会造成催化剂及高沸点溶剂损失严重。气相法工艺简单,反应连续,虽会产生积碳导致催化剂失活,但可通过再生使其活性恢复。催化剂的再生一般采用煅烧、通入水蒸气和用高沸点溶剂洗涤等方法[15-16]。
本工作采用沉淀法制备了β晶型的碱性Li3PO4催化剂[17],并对催化剂进行了表征。采用气相法研究了Li3PO4催化剂在PO异构化反应中的积碳及再生,考察了再生方法和条件对Li3PO4催化剂性能的影响,并探讨了Li3PO4催化剂失活与再生的机理。
1 实验部分
1.1 试剂
PO, LiOH·H2O, Na3PO4·12H2O:分析纯,成都市科龙化工试剂厂。
1.2 催化剂的制备
称取Na3PO4·12H2O(38.012 g)溶于150 mL的去离子水中(65 ℃),配成Na3PO4溶液备用。称取LiOH·H2O(16.784 g)溶于150 mL的去离子水中(65 ℃),将所得LiOH溶液滴加到Na3PO4溶液中,在65 ℃下搅拌1 h,陈化3 h,生成白色的Li3PO4沉淀。过滤、洗涤至滤液pH=12,在120 ℃下真空干燥8 h,再于马弗炉中320 ℃下煅烧8 h,研磨后得到碱性Li3PO4催化剂。
1.3 催化剂性能的评价
称取3份1.50 g的碱性Li3PO4催化剂分别装填于φ10 mm的U形反应管中。通过质量流量计调节氮气流量为5 mL/min;用计量泵控制液态PO的流量,使其WHSV=9 h-1。预热温度设为240 ℃;程序升温炉的温度分别设定为280,300,320 ℃;得到的产物在-6 ℃下冷凝。反应结束后,失活催化剂的质量分别为1.97,1.95,1.98 g。
1.4 催化剂的再生
1.4.1 煅烧法
称取失活催化剂1.97 g装入坩埚,置于马弗炉中400 ℃下煅烧8 h,得到再生的Li3PO4催化剂。
1.4.2 水蒸气洗涤法
称取失活催化剂1.95 g装入固定床反应器,通入水蒸气(预热器温度设为120 ℃,催化剂床层温度设为250 ℃),持续7 h,得到再生的Li3PO4催化剂。
1.5 表征和分析方法
XRD表征采用Bruker公司D8型X射线衍射仪,CuKα射线,波长0.154 05 nm。热重分析采用Mettler Toledo公司TGA/SDTA 851e型热重/差热同步分析仪,N2气氛,温度从50 ℃升至800 ℃,升温速率10 ℃/min。BET表征采用金埃普科技公司Vsorb-2800型比表面分析仪。FTIR和原位DRIFTS表征采用Thermo Scientific公司Nicolet iZ10型傅里叶变换红外光谱仪,波长范围650~4 000 cm-1,MCT检测器,原位DRIFTS实验中通入3 mL/min的N2保护气和3 mL/min的O2,测试温度350 ℃。MAS NMR表征采用Bruker公司Advance Ⅲ 400MHz型宽腔固体核磁共振谱仪。
PO异构化反应产物的分析采用北京北分瑞利分析仪器有限责任公司SP-1000型气相色谱仪,FF-AP毛细管色谱柱,柱长30 m,内径0.25 mm,气化温度230 ℃,检测温度190 ℃;柱温程序升温:50~120 ℃,10 ℃/min。
2 结果与讨论
2.1 催化剂的使用寿命
不同反应温度下Li3PO4催化剂性能随反应时间的变化见图1。由图1可见,随反应温度的升高,PO转化率和AA选择性均呈下降的趋势。实验中将PO转化率低于40%视为失活。在280 ℃下反应时,PO转化率和AA选择性均较高,且使用寿命较长(54.0 h),失活时的PO转化率为39.6%,AA选择性为84.8%。在300 ℃下反应时,催化剂使用寿命为51.0 h,失活时的PO转化率为38.6%,AA选择性为83.8%。在320 ℃下反应时,催化剂使用寿命为38.0 h,失活时的PO转化率为37.5%,AA选择性为86.6%。随反应温度的升高,催化剂使用寿命明显缩短,说明反应温度越高,表面积碳形成的速度越快,导致催化剂使用寿命缩短。

图1 不同反应温度下Li3PO4催化剂性能随反应时间的变化
2.2 再生后催化剂的性能
2.2.1 煅烧法
通过煅烧法对280 ℃下反应后的Li3PO4催化剂进行再生,并将再生后的催化剂重新装入固定床反应器中,进行PO异构化反应,实验结果见表1。
由表1可看出,再生后的Li3PO4催化剂上PO转化率基本能恢复到新鲜Li3PO4催化剂的水平,且颜色由灰色变为白色,与使用前的催化剂色泽一致;煅烧后催化剂的质量明显下降,由1.97 g降至1.52 g,这是由于失活催化剂表面的积碳高温分解所致。再生后催化剂的质量比新鲜催化剂(1.50 g)重0.02 g,可能是400 ℃下煅烧后有极少量积碳未分解。
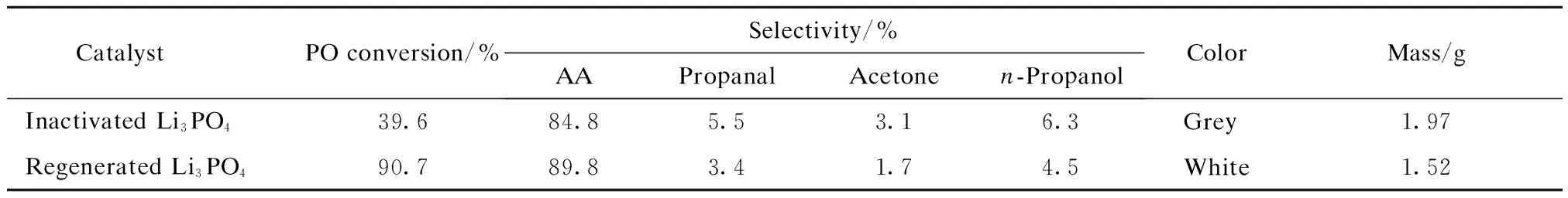
表1 煅烧法再生后Li3PO4催化剂的性能
Regeneration conditions: inactivated Li3PO4catalyst was calcined at 400 ℃ for 8 h.
Reaction conditions: 280 ℃, WHSV of PO 9 h-1.
2.2.2 水蒸气洗涤法
通过水蒸气洗涤法对300 ℃下反应后的Li3PO4催化剂进行再生,并将再生后的催化剂重新装入固定床反应器中,进行PO异构化反应,实验结果见表2。由表2可看出,再生后Li3PO4催化剂的颜色变浅,质量由1.95 g降至1.44 g,PO转化率恢复到80.7%。
水蒸气洗涤法使失活Li3PO4催化剂再生的原因可能有两种:1)通入过热水蒸气进行原位吹扫,从而除去积碳,是物理过程;2)在高温环境中水蒸气与积碳有机物进行化学反应,从而除去积碳。再生Li3PO4催化剂的质量比新鲜Li3PO4催化剂的质量减少,是由于清洗积碳的同时少量催化剂被损失,具体机理需进一步探究。
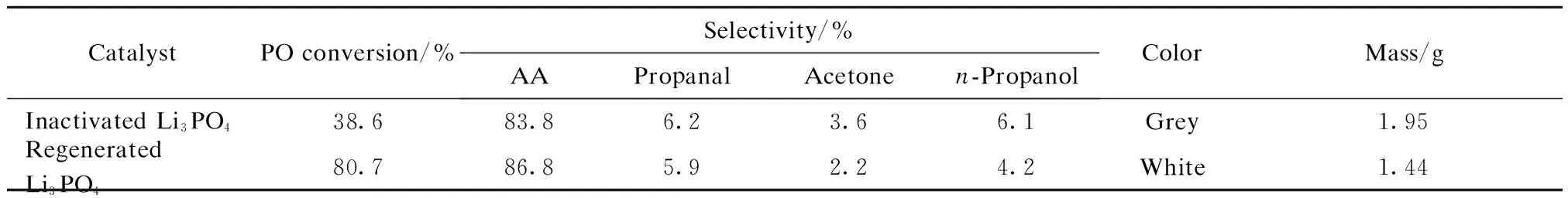
表2 水蒸气洗涤法再生后Li3PO4催化剂的性能
Reaction conditions:inactivated Li3PO4catalyst was washed with steam at 250 ℃ for 7 h.
Reaction conditions: 300 ℃, WHSV of PO 9 h-1.
2.3 XRD表征结果
新鲜的、失活的及再生后Li3PO4催化剂的XRD谱图见图2。

图2 新鲜的(a)、失活的(b)及再生后(c) Li3PO4催化剂的XRD谱图
由图2中(110),(101),(210)晶面的几个主要特征峰可判断出,Li3PO4催化剂的晶型是β-Li3PO4(JCPDS No. 25-1030),且峰形较尖锐,说明结晶度较高。在各阶段Li3PO4催化剂的晶型均不改变,且无新物相生成。失活Li3PO4催化剂的峰强度比新鲜Li3PO4催化剂的弱,其原因可能是失活Li3PO4催化剂表面覆盖了大量积碳,使其峰强度减弱。XRD表征结果显示,在整个过程中,Li3PO4催化剂的结构及晶型均未改变,证明了再生方法的可行性。
2.4 TG分析结果
新鲜的、失活的及再生后Li3PO4催化剂的TG曲线见图3。由图3可看出,新鲜Li3PO4催化剂在50~600 ℃之间的总失重达3.2%,50~300 ℃为第一阶段,约有0.9%的失重,主要是吸附水的脱附;300~600 ℃为第二阶段,有2.3%的失重,是碱性Li3PO4催化剂表面的CO32-分解和少量结晶水失去引起的。而失活Li3PO4催化剂在整个过程中的总失重高达23%,且在300~500 ℃之间的失重峰斜率最大,失重过程几乎都在此阶段完成,失重22%,说明是由Li3PO4催化剂表面的积碳在该温度区间发生分解引起的;在300 ℃以下及500 ℃以上的失重基本与新鲜催化剂相同,失重原因也是吸附水脱附及CO32-分解;在600 ℃以上失重现象几乎不发生,且失活催化剂的颜色由灰黑色变成白色,与再生后的催化剂颜色相同。再生催化剂与新鲜催化剂的失重趋势基本一样,甚至失重趋向于零,说明再生Li3PO4催化剂表面积碳去除较彻底;且由于采用煅烧法再生,再生Li3PO4催化剂中的吸附水和结晶水含量均大幅降低。
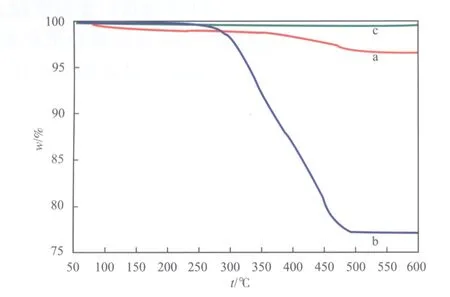
图3 新鲜的(a)、失活的(b)及再生后(c) Li3PO4催化剂的TG曲线
2.5 BET分析结果
催化剂积碳失活机理主要分为两种:活性位被覆盖和孔道被堵塞[18]。新鲜的、失活的及再生后Li3PO4催化剂的织构性质见表3。由表3可知,失活Li3PO4催化剂的比表面积比新鲜Li3PO4催化剂降低了约90%,同时PO转化率下降明显,说明Li3PO4催化剂的失活是由于积碳覆盖在催化剂表面,使其比表面积减小、孔隙率降低、活性中心减少等原因造成的。

表3 新鲜的、失活的及再生后Li3PO4催化剂的织构性质
SBET,V: specific surface area and pore volume calculated by the BET method, respectively.
新鲜的、失活的及再生后Li3PO4催化剂的N2吸附-脱附等温线及孔分布曲线见图4。由图4可知,Li3PO4催化剂的吸附等温线是Ⅳ型等温线,H3型回滞环,说明Li3PO4催化剂是中孔结构,且孔结构很不规整。新鲜的和再生后Li3PO4催化剂的最可几孔径约在5 nm,而失活Li3PO4催化剂的最可几孔径明显减小,说明失活催化剂表面覆盖积碳,使其孔径变小甚至完全堵塞。再生Li3PO4催化剂由于积碳被分解,比表面积和孔径几乎恢复到新鲜催化剂的状态。
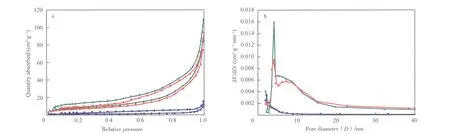
图4 新鲜的、失活的及再生后Li3PO4催化剂的N2吸附-脱附等温线及孔分布曲线
2.6 FTIR表征结果
无机盐的FTIR谱图中只有酸根离子具有特征峰,在Sadler标准FTIR谱图中,PO43-的吸收峰在950~1 200 cm-1处,且峰强度较强;正磷酸盐在600 cm-1附近还存在一个强度稍弱的特征峰。新鲜和失活Li3PO4催化剂的FTIR谱图见图5。由图5可见,新鲜的及失活的Li3PO4催化剂均在1 056 cm-1附近出现强吸收峰,证明了PO43-的存在;在595 cm-1附近出现弱吸收峰,证明了Li3PO4的存在。与新鲜催化剂明显不同的是,失活催化剂的FTIR谱图中在2 848~2 988 cm-1处出现吸收峰,这是饱和结构—CH3和—CH2的伸缩振动峰,说明失活催化剂表面积碳为含氢有机化合物[19]。此外,FTIR谱图中在3 470 cm-1处的吸收峰归属于H2O或碱性环境的OH-,失活前后的催化剂在该处的吸收峰强度相似,说明Li3PO4的化学环境基本不变。2 356 cm-1处的吸收峰归属于CO2的伸缩振动;1 445 cm-1处的吸收峰归属于CO32-的伸缩振动。与失活催化剂相比,新鲜催化剂在1 445 cm-1处的吸收峰强度更强,这是因为新鲜催化剂中的CO32-含量更高,而在使用过程中CO32-逐渐分解导致失活催化剂中的CO32-含量降低。
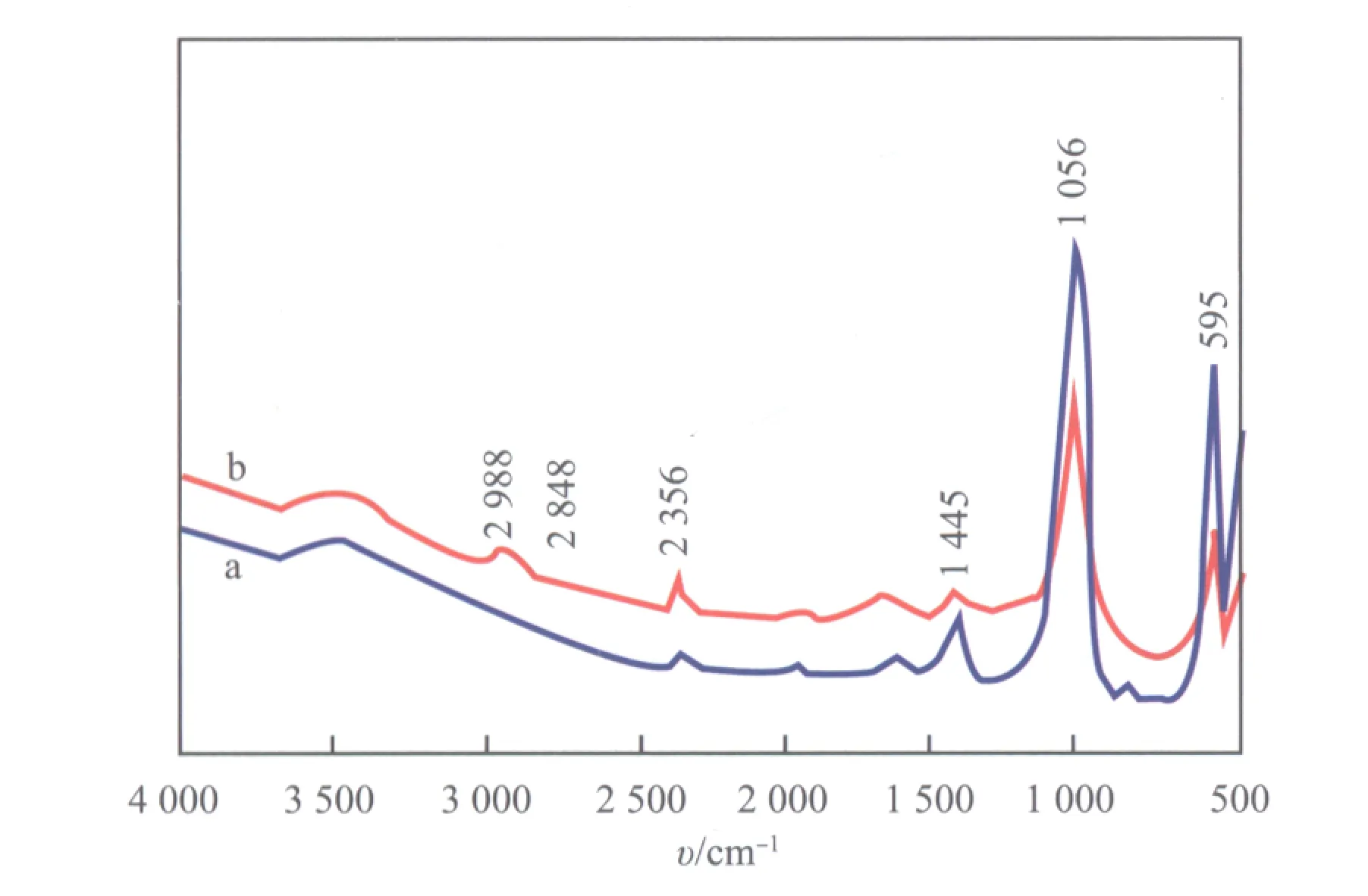
图5 新鲜的(a)和失活的(b) Li3PO4催化剂的FTIR谱图
采用原位DRIFT方法表征了失活Li3PO4催化剂在350 ℃下的积碳分解情况,表征结果见图6。
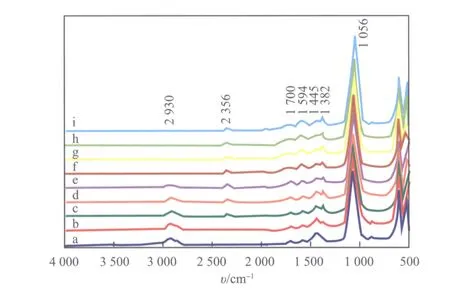
图6 在O2 和 N2气氛下,经350 ℃加热的失活Li3PO4催化剂在不同时间下的原位DRIFT谱图
由图6可看出,随时间的延长,2 930 cm-1处有机物C—H键的伸缩振动峰强度逐渐减弱,约50 min时基本消失,说明在350 ℃下积碳分解所需时间为40~50 min;2 356 cm-1处CO2吸收峰的变化说明分解过程中释放出气体CO2,在1 min和10 min时 CO2的累积释放量不大,从20 min开始出现明显的CO2吸收峰,50 min后该吸收峰强度逐渐减弱,这是因为产生的气体量逐渐减少且被N2保护气带走;1 445 cm-1处CO32-的吸收峰强度也逐渐减弱,应是CO32-的逐渐分解所致。
2.7 MAS NMR表征结果
2.7.11H NMR表征结果
新鲜和失活Li3PO4催化剂的1H NMR谱图见图7。由图7可见,新鲜Li3PO4催化剂只在化学位移(δ)为4.673处出现一个单峰,归属于碱性Li3PO4催化剂中的OH-;而失活催化剂因积碳覆盖该峰位移至δ=4.868处,并在δ=0.860,0.369处出现了两组新峰,分别归属于饱和烷烃中的氢核。
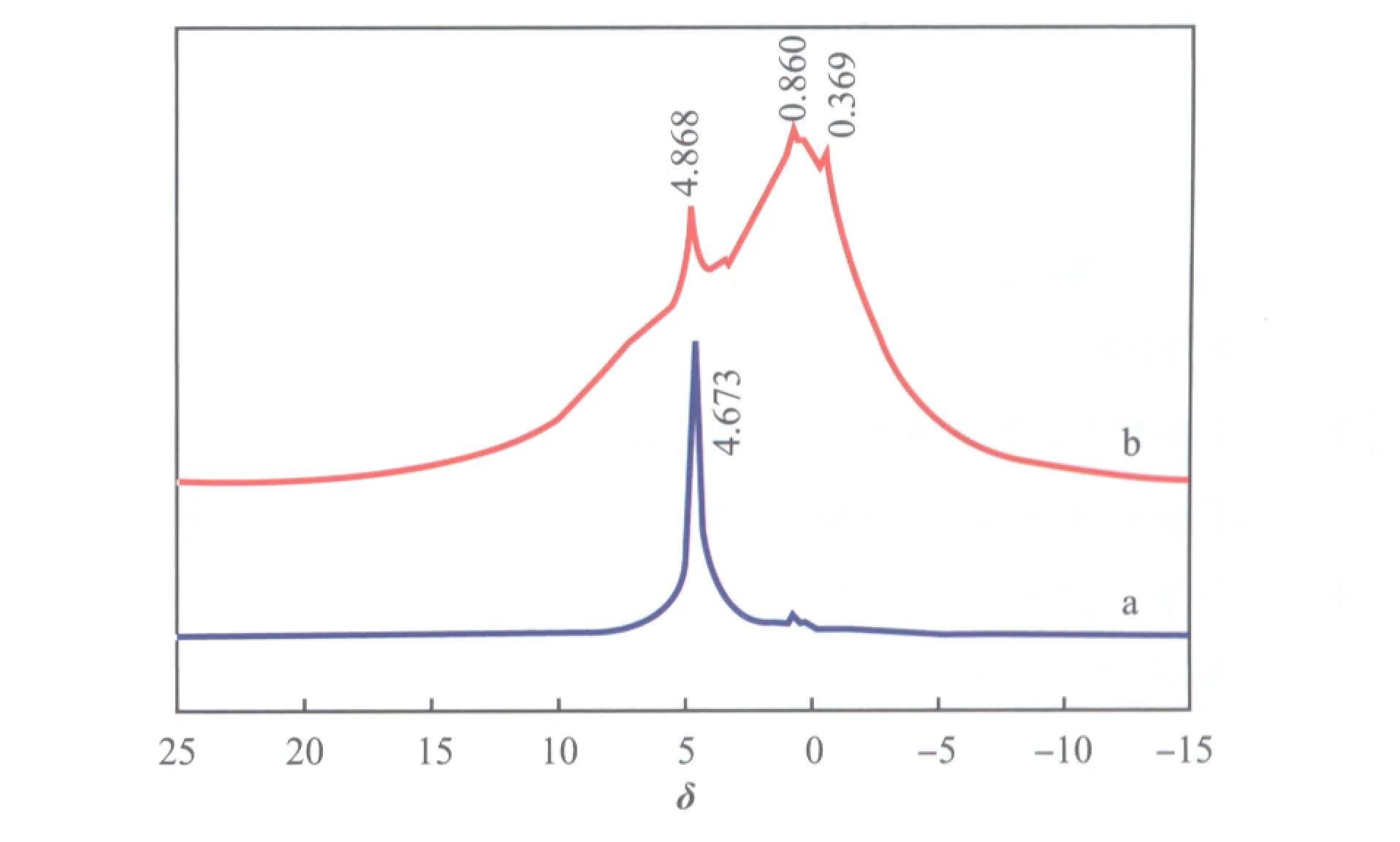
图7 新鲜的(a)和失活的(b) Li3PO4催化剂的1H NMR谱图
2.7.27Li NMR表征结果
新鲜的和失活的Li3PO4催化剂的7Li NMR谱图见图8。从图8可看出,两种催化剂的谱图基本相同,说明失活催化剂中Li的化学环境并未改变。

图8 新鲜的(a)和失活的(b) Li3PO4催化剂的7Li NMR谱图
3 结论
1)采用沉淀法制备的碱性Li3PO4催化剂应用于PO异构化制烯丙醇反应,在280 ℃下的使用寿命最长,且PO转化率和AA选择性也相对较高。
2)碱性Li3PO4催化剂失活的主要原因是表面积碳,可采用煅烧法和水蒸气洗涤法进行再生,从而延长催化剂的使用寿命。
3)由TG分析结果可知,失活Li3PO4催化剂在300~500 ℃内的失重率达22%,说明积碳在该温度内分解,故在400 ℃下煅烧,再生效果较好,再生后Li3PO4催化剂的活性与新鲜Li3PO4催化剂相近。
[1] Charles K, Alan M, Floyd M, et al. The Crystal Structure of Twinned Low-Temperature Lithium Phosphate[J].InorgChem, 1967, 6(1): 119-125.
[2] Du Yaojun A, Holzwarth N A W. Mechanisms of Li+Diffusion in Crystalline γ- and β-Li3PO4Electrolytes from First Principles[J].PhysRevB:CondensMatter, 2007, 76(17):174302-174316.
[3] Liu H C, Yen S K. Electrolytic Li3PO4Coating on Pt[J].JPowerSources, 2006, 159(1): 245-248.
[4] 牛锛, 满丽莹, 齐恩磊, 等. 磷酸锂粉体的制备与表征[J]. 硫磷设计与粉体工程, 2011(2): 27-28.
[5] Zhao Shixi, Ding Hao, Wang Yanchao, et al. Improving Rate Performance of LiFePO4Cathode Materials by Hybrid Coating of Nano-Li3PO4and Carbon[J].JAlloysCompd, 2013, 566: 206-211.
[6] 胡意,艾常春,刘洋,等. 紊流循环法合成超细磷酸锂及表征[J]. 化工学报, 2014, 65(3): 1099-1103.
[7] Chaehwan J, Ho-Gun S, DuckRye C, et al. Fabrication of the
Planar-Type CO2Gas Sensor Using an Evaporated Li3PO4Film and Its Sensing Characteristics[J].MetMaterInt, 2009, 15(1): 101-105.
[8] Zhang Weixin, Chen Lingling, Yang Zeheng, et al. An Optical Humidity Sensor Based on Li3PO4Hollow Nanospheres[J].SensActuators,B, 2011, 155(1): 226-231.
[9] Marzouk M A, ElBatal H A, Abdel Ghany A M, et al. Ultraviolet, Visible, ESR, and Infrared Spectroscopic Studies of CeO2-Doped Lithium Phosphate Glasses and Effect of Gamma Irradiation[J].JMolStruct, 2011, 997(1/3): 94-102.
[10] Mascaraque N, Tricot G, Revel B, et al. Nitrogen and Fluorine Anionic Substitution in Lithium Phosphate Glasses[J].SolidStateIonics, 2014, 254: 40-47.
[11] 马卫华, 陆路德, 杨绪杰, 等. 环氧丙烷催化异构化及动力学研究[J]. 化学反应工程与工艺, 2005, 21(5): 397-401.
[12] Guo Xianwei, Fang Xiangpeng, Mao Ya, et al. Capacitive Energy Storage on Fe/Li3PO4Grain Boundaries[J].JPhysChemC, 2011, 115(9): 3803-3808.
[13] Stamicarbon B V Corporation. Process for Preparing a Basic Lithium Phosphate Catalyst for the Isomerization of Alkene Oxides : US, 4720598 [P]. 1988-01-19.
[14] Daicel Chem Ind Ltd. Preparation of Allyl Alcohol from Propylene Oxide: JP, 01272539[P]. 1989-10-31.
[15] Huch G L. Process for Regenerating Li3PO4Catalyst: RO, 90897[P]. 1987-04-27.
[16]Deutsche Gold-Und Silber-Scheideanstalt Vormals Roessler. Production of an Alkenol from an Alkene Oxides: US, 4065510[P]. 1977-12-27.
[17] Arco Chemical Technology, LP. Preparation of Lithium Phosphate Catalysts: US, 6803491[P]. 2004-10-12.
[18] 周丽雯, 王飞, 罗漫, 等. ZSM-5催化剂在乙醇脱水反应中的失活与再生[J]. 石油化工, 2008, 37(4): 333-337.
[19] 高鸿宾. 有机化学[M]. 4版. 北京:高等教育出版社,2005, 1: 298.
(编辑 安 静)
Deactivation and Regeneration of Basic Lithium Phosphate Catalyst
LiHao,LiFei,ZhouXuan,MaWeihua,ZhongQin
(DepartmentofChemicalEngineering,NanjingUniversityofScienceandTechnology,NanjingJiangsu210094,China)
The deactivation and regeneration of basic lithium phosphate catalyst used in the isomerization of propylene oxide to allyl alcohol were studied. The results showed that the basic lithium phosphate catalyst deactivated after the reaction at 280 ℃ for 54 h, which was due to carbon deposition containing H element. The carbon deposition could be removed by washing with steam or calcining. The fresh, deactivated and regenerated catalysts were characterized by means of XRD, FTIR, in-situ DRIFT, BET, TG and MAS NMR. The specific surface area and pore volume of the deactivated catalyst decreased without the formation of new phase. The decomposition temperature of the carbon deposition was in the range of 300-500 ℃ and, at 350 ℃ the decomposition needed 40-50 min. The conversion of propylene oxide and the selectivity to allyl alcohol on the catalyst regenerated by the calcining reached 90.7% and 89.8%, respectively, similar to those on the fresh catalyst.
propylene oxide; isomerization;allyl alcohol;lithium phosphate catalyst; carbon deposition; deactivation; regeneration
2014-09-22;[修改稿日期] 2014-12-26。
李浩(1989—),男,江苏省南通市人,硕士生。联系人:马卫华,电话 025-84315517,电邮 maweihuacn@163.com。
国家自然科学基金项目(21276127;51106076)。
1000-8144(2015)04-0453-06
TQ 426
A
猜你喜欢
发明与创新·大科技(2019年6期)2019-09-06
石油石化绿色低碳(2019年6期)2019-01-14
汽车观察(2018年10期)2018-11-06
——会偷偷侵蚀你的发动机!
人民交通(2016年8期)2017-01-05
柴油机设计与制造(2015年3期)2015-12-05
化工进展(2015年3期)2015-11-11
华东理工大学学报(自然科学版)(2015年3期)2015-11-07
河南科技(2015年2期)2015-02-27
中国粮油学报(2014年8期)2014-02-06
微创医学(2012年6期)2012-01-13
