
一测多评法测定板蓝根颗粒中4种成分的含量
2015-07-07 16:05:24池絮影崔曰新张蜀邓红
中国生化药物杂志 2015年10期
池絮影,崔曰新,张蜀,邓红
(广东药学院 药物研究所/广东省药物新剂型重点实验室,广东 广州 510006)
一测多评法测定板蓝根颗粒中4种成分的含量
池絮影,崔曰新,张蜀,邓红Δ
(广东药学院 药物研究所/广东省药物新剂型重点实验室,广东 广州 510006)
目的 建立同时测定板蓝根颗粒中腺苷、尿苷、鸟苷、(R,S)-告依春的一测多评法。方法 以腺苷为内标物,确定板蓝根中其他3种成分相对于腺苷的校正因子,通过相对校正因子(relative correction factor,RCF)对尿苷、鸟苷、(R,S)-告依春进行定量,实现一测多评(计算法);同时采用外标法测定板蓝根颗粒中4种成分的含量(实测值),并比较一测多评法与外标法测定结果的差异。结果 各成分相对校正因子重现性良好。尿苷、鸟苷、(R,S)-告依春按一测多评方法与外标法测定结果差异无统计学意义。结论 本研究建立的一测多评法简便,测定结果准确可靠,可为板蓝根颗粒的质量控制提供参考。
一测多评;腺苷;尿苷;鸟苷;(R,S)-告依春;UPLC;HPLC
板蓝根颗粒是国家基本药物,收载于《中国药典》2010年版一部,为板蓝根经水提醇沉等步骤制成的单方中药颗粒剂,现行标准无定量控制指标[1],而《中国药典》2010年版一部收载的板蓝根[2]仅使用(R,S)-告依春单一检测指标。按照传统中医药学的“整体观”,中药药效是药品中多个药效成分的综合效应[3-4],因此有必要建立板蓝根颗粒的多成分同步质量控制方法。然而现实中的对照品供需矛盾和多指标质控高昂的检测成本又限制了多指标质量控制模式在实际生产、科研、监督中的应用。一测多评技术(quantitative analysis of multi-components by single-marker,QAMS)是当前解决这一矛盾的重要途径之一[5-7]。本研究以相对稳定且对照品廉价易得的腺苷为内标物,考察建立一测多评法测定板蓝根颗粒中尿苷、鸟苷、(R,S)-告依春的量的可行性,以期为板蓝根颗粒质量标准的提高提供充分的实验依据,实现板蓝根颗粒的多成分质量评价。
1 材料与方法
1.1 材料
1.1.1 仪器 :超高效液相色谱仪(美国Waters公司);高效液相色谱仪(美国Dionex公司);分析柱为phenomen Kinetex(100×4.6 mm, 2.6 μm);XSELECTM HSS T3(100×2.1 mm,2.5 μm);Ecosil C18(250×4.6 mm,5 μm)。CP225D型电子分析天平(德国Sartorius公司);BS224S型电子分析天平(德国Sartorius公司)。
1.1.2 药品与试剂:板蓝根颗粒(批号见表5,规格为3 mg/袋);(R,S)-告依春,腺苷对照品(批号111753-201304、110876-200204,中国食品药品检定研究所);尿苷对照品(批号8-SCC-56-1,Toronto Research Chemicals Inc.);鸟苷对照品(批号LE20O31,北京百灵威科技有限公司);甲醇(HPLC级,Merck),水(屈臣氏蒸馏水),其他试剂均为分析纯。
1.2 方法
1.2.1 溶液的制备
① 混合对照品溶液的制备:取尿苷对照品、鸟苷对照品各约10 mg、(R,S)-告依春对照品约20 mg及腺苷对照品约12.5 mg,精密称定,置50 mL量瓶中,用甲醇溶解并稀释至刻度,摇匀,即得混合对照品贮备液;精密量取上述溶液1 mL,置10 mL量瓶中,加水稀释至刻度,摇匀,滤过,即得。
② 板蓝根颗粒供试品溶液的制备:取板蓝根颗粒,研细,取约0.67 g,精密称定,置具塞锥形瓶中,精密加入5%甲醇10 mL,密塞,称定重量,超声5 min,放冷,再称定重量,用5%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
③ 阴性对照溶液的制备:按板蓝根颗粒制备方法,制备不含板蓝根药材的阴性样品,并按供试品溶液配制方法制成阴性对照溶液。
1.2.2 专属性试验:分别取“1.2.1”项下的混合对照品溶液和供试品溶液,按“②”和“③”项下色谱条件进样检测,记录色谱图。
1.2.3 线性关系的考察:精密量取“1.2.1”项下①混合对照品贮备液,分别加水稀释,摇匀,配制成尿苷浓度为2.058、4.116、10.29、15.44、20.58、30.87、51.45、102.9 μg/mL,鸟苷浓度为2.070、4.140、10.35、15.53、20.70、31.05、51.75、103.5 μg/mL,(R,S)-告依春浓度为4.086、8.172、20.43、30.65、40.86、61.29、102.2、204.3 μg/mL,腺苷浓度为2.506、5.012、12.53、18.80、25.06、37.59、62.65、125.3 μg/mL的系列对照品溶液。在“②”项下色谱条件下,分别进样检测,记录色谱图。
1.2.4 一测多评法方法学考察
① 一测多评法原理[8-11]:在一定的范围内(线性范围内)成分的量(质量或浓度)与检测器响应成正比,即:W=f·A。在中药多指标质量评价时,以待测组分中某一成分(有对照品供应者)为内标,建立该组分与其他组分之间的相对校正因子(relative correction factor, RCF),通过校正因子计算其他组分的含量。
校正因子f=(As/Cs)/(Ar/Cr)
①
式中As为内标物对照品峰面积;Cs为内标物浓度;Ar为某待测成分对照品峰面积;Cr为某待测成分对照品浓度。
含量Cx=f×Ax/(As/Cs)
②
式中Cx为供试品中某待测成分浓度;f为内标物s对待测成分x的校正因子;Ax为供试品中待测成分x的峰面积;As为供试品中内标物s的峰面积;Cs为供试品中内标物s的浓度;Ax为供试品中待测成分x的峰面积。
② UPLC色谱条件:色谱柱为phenomen Kinetex (100 mm×4.6 mm, 2.6 μm),流动相为甲醇-水,梯度洗脱(0~3 min,1%~3%甲醇;3~11 min,3%~5%甲醇;11~13 min,5%~70%甲醇;13~13.5 min,70%~1%甲醇;13.5~16 min,1%~1%甲醇)。流速为0.600 mL/min;柱温为30 ℃;检测波长为254%nm;进样量为2 μL。
③ HPLC色谱条件:色谱柱为Ecosil C18(250 mm×4.6 mm,5 μm),流动相为甲醇-水,梯度洗脱(0~3 min,3%~3%甲醇;3~20 min,3%~10%甲醇;20~40 min,10%~70%甲醇;40~50 min,70%~70%甲醇;50.01~60 min,3%~3%甲醇)。流速为0.800 mL/min;柱温为30 ℃;检测波长为254 nm;进样量为20 μL。
1.2.5 稳定性考察:取同一供试品溶液,分别于0、2、4、8、12 h进样测定,计算RSD值。
1.2.6 精密度试验:取同一供试品溶液,连续重复进样6次,记录峰面积,计算RSD值。
1.2.7 重复性试验:精密称取同一批号板蓝根颗粒(批号:140812-2),按“1.2.1”项下平行制备供试品溶液6份,依法测定。
1.2.8 加样回收率试验:取已知含量的板蓝根颗粒(批号:140812-2),研细,取约0.34 g,精密称定6份,准确加入含一定量的尿苷、鸟苷、(R,S)-告依春和腺苷的混合对照溶液,按照“1.2.1”项下方法制备供试液,依法进样,测定含量。
1.2.9 一测多评法与外标法测定结果的比较:精密称取板蓝根颗粒,按“1.2.1”项下制备供试品溶液,依法测定,用外标法(external standard method,ESM)对4个待测成分定量测定,与一测多评法测定结果比较,验证一测多评法用于板蓝根颗粒中4种成分测定的准确性。
2 结果
2.1 专属性试验 尿苷、鸟苷、(R,S)-告依春和腺苷的分离度良好,供试品溶液中各待测成分峰不受其他峰干扰。
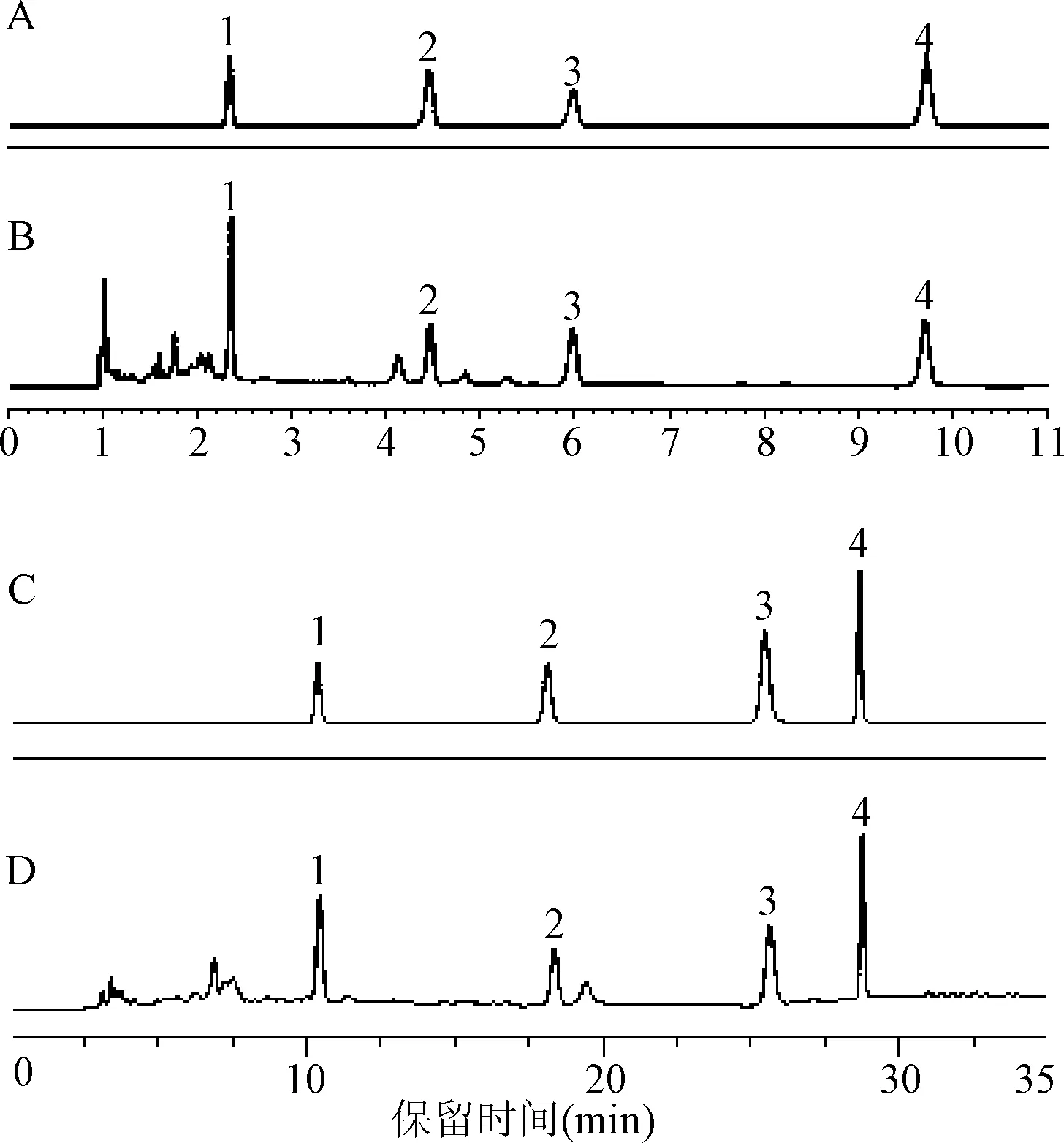
图1 混合对照品和供试品的UPLC及HPLC图1.尿苷;2.鸟苷;3.(R,S)-告依春;4.腺苷A:混合对照品(UPLC);B:供试品(UPLC); C:混合对照品(HPLC);D:供试品(HPLC)Fig.1 Comparison of UPLC and HPLC Chromatograms of mixed reference substances and samples1.uridine;2.guanosine;3.(R,S)-goitrin;3.adenosineA: Mixed standard (UPLC) B: The test (UPLC) C: Mixed standard (HPLC) D: The test (HPLC)
2.2 线性关系考察 以对照品质量浓度(μg/mL)为横坐标,峰面积(A)为纵坐标,绘制标准曲线,结果见表1。
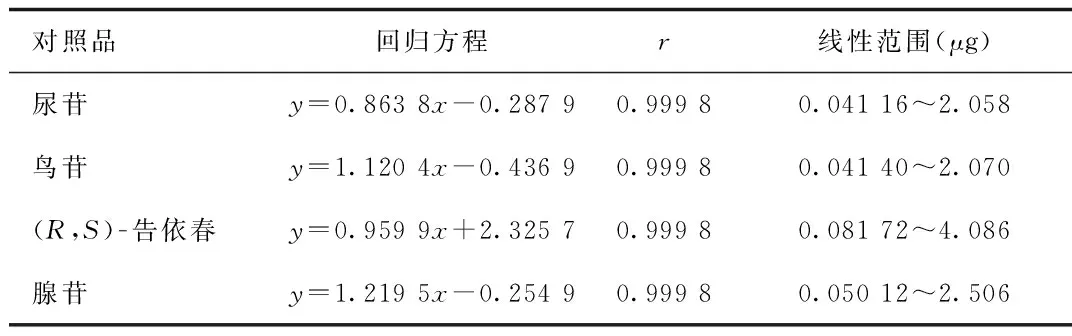
表1 线性回归曲线
2.3 相对校正因子和相对保留时间测定
2.3.1 校正因子计算:在建立一测多评法对板蓝根颗粒进行质量评价时,根据斜率校正法[12-14],以腺苷为内标物,将各待测组分标准曲线的截距校正为0后,得各待测组分经校正后的标准曲线回归方程A=kc,由公式①得
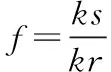
③
式中ks为腺苷对照品经校正后的回归方程的系数;kr为其他待测成分对照品经校正后的回归方程的系数。

表2 校正因子
2.3.2 校正因子的耐用性考察[15]:本研究通过测定校正因子在不同品牌仪器、不同规格色谱柱、不同流速、不同柱温的重现性,考察其耐用性。结果显示,校正因子的耐用性良好。见表3。
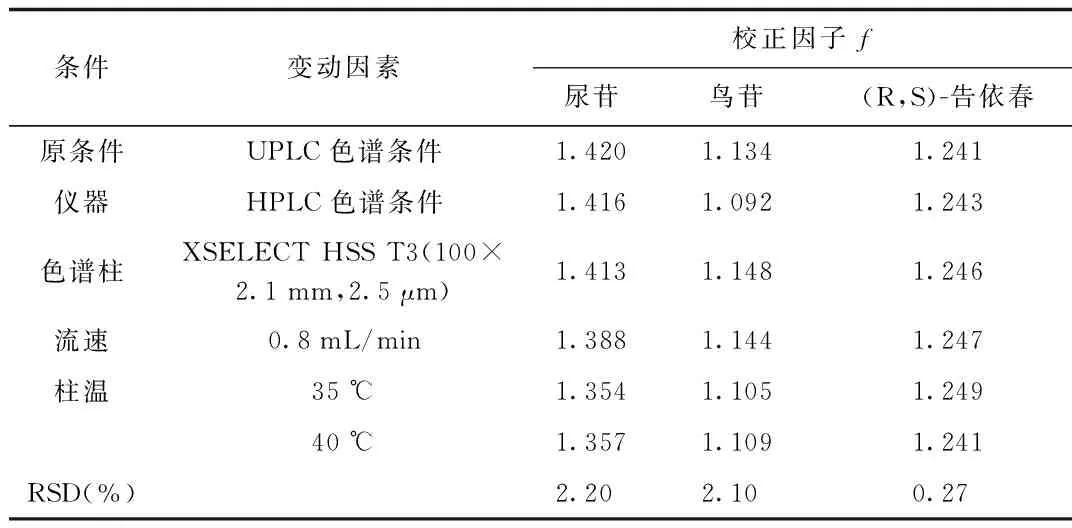
表3 校正因子的耐用性试验
2.3.3 待测组分色谱峰的定位:腺苷通过对照品定位,其他待测组分采用相对腺苷保留时间(relative retention time;RRT值)进行定位。使用一测多评法进行含量测定时,通过腺苷峰的保留时间计算其他待测成分峰的RRT值,根据RRT值及光谱吸收可对尿苷、鸟苷及(R,S)-告依春准确定位。本实验测定了相对保留值在不同品牌仪器、不同规格色谱柱、不同流速、不同柱温的重现性,结果尿苷、鸟苷、(R,S)-告依春的相对保留时间分别为0.2476、0.4488、0.6363,RSD均<5%。
2.4 稳定性考察 在室温条件下,峰面积无明显变化,供试品溶液中尿苷、鸟苷、(R,S)-告依春和腺苷的RSD分别为0.071%,0.17%,0.75%和0.30%,表明供试品溶液在室温12 h内稳定。
2.5 精密度试验 供试品中尿苷峰面积的RSD为0.067%,鸟苷峰面积的RSD为1.2%,(R,S)-告依春峰面积的RSD为0.076%,腺苷峰面积的RSD为0.34%,表明仪器精密度良好。
2.6 重复性试验 供试品中尿苷、鸟苷、(R,S)-告依春和腺苷4种成分含量平均值分别为1.519 mg/袋,0.7775 mg/袋,1.699 mg/袋,1.220 mg/袋,RSD分别为1.5%,1.9%,0.41%,0.60%,表明该方法重复性良好。
2.7 加样回收率试验 尿苷的平均回收率为99.7%(RSD=0.39%),鸟苷的平均回收率为102.4%(RSD=0.50%),(R,S)-告依春的平均回收率为99.7%(RSD=0.89%),腺苷的平均回收率为101.1%(RSD=0.34%),表明该方法的准确度良好。
2.8 一测多评法与外标法测定结果的比较 结果表明2种方法所得含量无显著性差异(相对偏差<2%),说明建立的一测多评法可用于板蓝根及其颗粒剂的多成分质量评价研究。见表4。
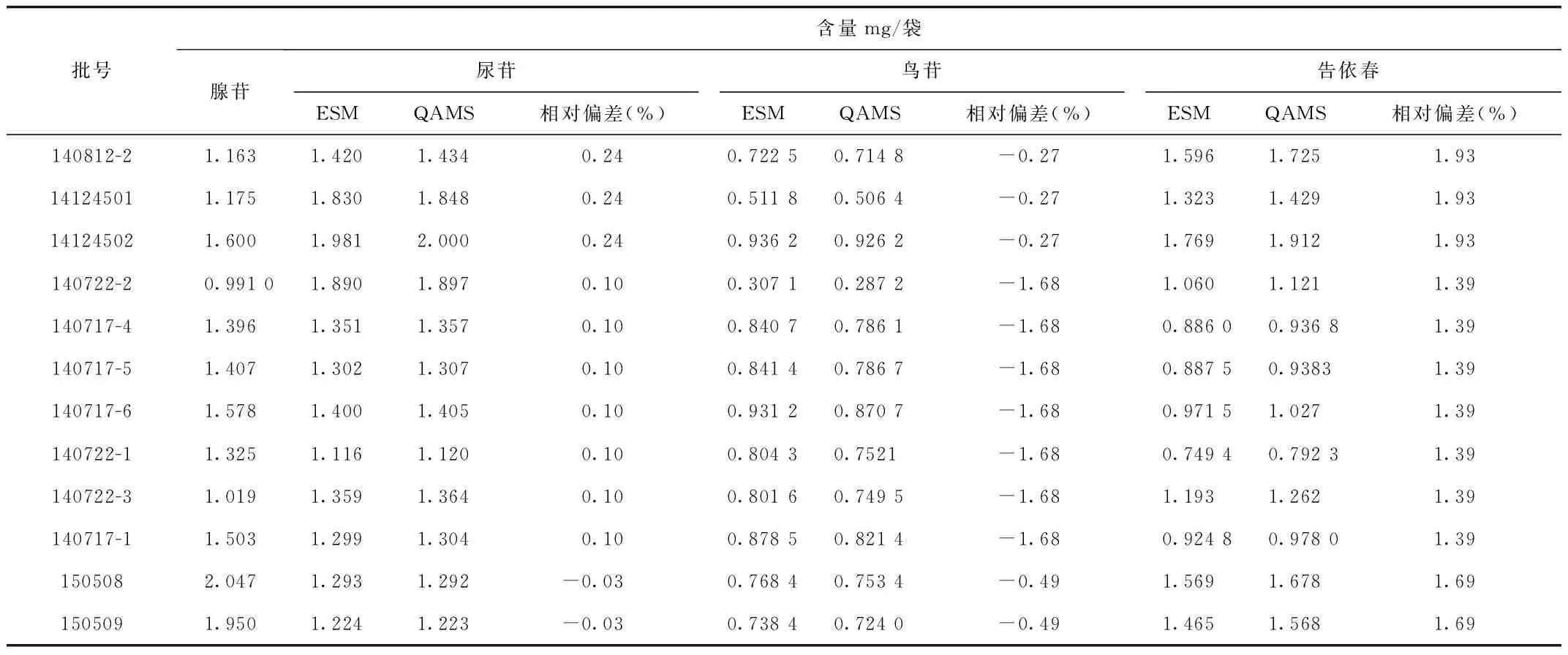
表4 一测多评法与外标法测定板蓝根颗粒中4种成分的含量
3 讨论
3.1 内标物的选择 板蓝根颗粒中的核苷类成分中较难得到,只有腺苷购自中国食品药品检定研究所,而较于(R,S)-告依春,腺苷较稳定且廉价易得,故选其为内标物,能更好地体现QAMS法简便、易操作、低成本的特点。
3.2 本文采用高分离效率、高灵敏度和高分辨率的UPLC色谱分离系统,10 min即可实现4个目标成分的分离,方便快捷,大大缩短了分析检测时间。同时考察了HPLC色谱体系,结果表明在不同色谱分析体系中,相对校正因子及相对保留时间的重现性良好,可用于方法的互相转换。
本研究初步建立了一测多评法用于板蓝根颗粒中4种成分的同步测定,与外标法实测值无显著性差异(相对偏差<2%)),说明建立的不同成分之间的校正因子具有较高的可信度,与外标法相比,能较大程度地节约多成分含量测定的检测成本,并为板蓝根药材及制剂的多指标质量控制模式提供新方法。
[1] 国家药典委员会.中国人民共和国药典(2010年版第二增补本)[S].北京:中国医药科技出版社, 2013: 132.
[2] 国家药典委员会.中国人民共和国药典(一部)[S].北京:中国医药科技出版社,2010:191.
[3] 邱咏薇,杨怀瑾.中医药的特色——整体观[J].河北中医,2004,26(12):945.
[4] Qiu J.Traditional medicine: a culture in the balance[J].Nature,2007,448:126-128.
[5] 王智民,高慧敏,付雪涛,等.“一测多评”法中药质量评价模式方法学研究[J].中国中药杂志,2006(23):1925-1928.
[6] 陆兔林,石上梅,蔡宝昌,等.基于一测多评的中药多成分定量研究进展[J].中草药,2012,43(12):2525-2529.
[7] 文乾映,龙芳,杨华,等.中药质量控制中一测多评法的应用进展[J].中国药房,2014,25(23):2185-2188.
[8] Zhu JJ,Wang ZM, Ma XY, et al.A quantitative method for simultaneous determination of four anthraquinones with one marker in Rhei Radix et Rhizoma [J].Chin Herb Med, 2012, 4(2): 157-163.
[9] 王智民,钱忠直,张启伟,等.一测多评法建立的技术指南[J].中国中药杂志,2011(6): 657-658.
[10] 逄瑜,孙磊,金红宇,等.替代对照品法在中药多指标含量测定中的应用与技术要求探讨[J].药物分析杂志,2013,33(1):169-177.
[11] 高慧敏,宋宗华,王智民,等.适合中药特点的质量评价模式-QAMS研究概述[J].中国中药杂志,2012,37(4):405-416.
[12] 何兵,杨世艳,张燕.一测多评中待测成分校正和定位的新方法研究[J].药学学报, 2012(12):1653-1659.
[13] Zhu JJ,Wang ZM,Kuang YH,et al.A quantitative method using one marker for simulataneous assay of ginsenosides in Panax ginseng and P.notoginseng [J].Acta Pharm Sin,2008,43:1211-1216.
[14] 赵一懿,郭洪祝,陈有根,等.中药多组分含量测定中相对校正因子计算方法的比较与建议[J].中国药品标准,2014,15(4):245-251.
[15] 冯伟红,杨菲,王智民,等.不同色谱条件对QAMS相对校正因子及相对保留值影响的实验研究[J].2012,37(21):3264-3267.
(编校:谭玲)
Quantitative analysis of 4 components in Isatidis Radix granules using single marker by QAMS
CHI Xu-ying, CUI Yue-xin, ZHANG Shu, DENG HongΔ
(Institute of Materia Medica, Guangdong Pharmaceutical University/Guangdong Provincial Key Laboratory of Advanced Drug Delivery, Guangzhou 510006, China)
ObjectiveTo establish a quantitative analysis of multi-components by single-marker (QAMS) for determination of four components (adenosine, uridine, guanosine, (R,S)-goitrin) in Isatidis Radix granules.MethodThe QAMS for Isatidis Radix granules was established and validated and adenosine was selected as the internal reference substance.The relative correction factor(RCF)of other three components were calculated.The contents of four components were determined by both external standard method and QAMS.The QAMS method was validated through comparison of the results obtained by the two different methods.ResultsThe RCFs of uridine, guanosine and (R, S)-goitrin all showed good reproducibility within certain ranges.For Isatidis Radix granules, uridine, guanosine and (R, S)-goitrin, there was no significant difference between the quantitative results of the two methods.ConclusionsThe QAMS method is simple, feasible and credible to be used for comprehensive quality control of Isatidis Radix granules.
QAMS; adenosine; uridine; guanosine; (R, S)-goitrin; UPLC; HPLC
池絮影,女,硕士在读,研究方向:中药制剂研究与开发,E-mail:cxysheen@163.com;邓红,通信作者,女,本科,教授级高级工程师,研究方向:中药新剂型和质量标准研究,E-mail:dengh361@sohu.com。
R927.2
A
1005-1678(2015)10-0137-04
猜你喜欢
检察风云(2022年5期)2022-04-05 13:42:39
世界科学技术-中医药现代化(2021年9期)2021-12-31 03:31:24
中国调味品(2018年7期)2018-07-17 02:08:48
益寿宝典(2018年35期)2018-01-26 17:00:09
中成药(2017年12期)2018-01-19 02:06:48
中国中医药信息杂志(2016年9期)2016-12-03 11:07:02
国外医药(抗生素分册)(2016年3期)2016-07-12 14:25:18
山东医药(2015年16期)2016-01-12 00:40:08
应用化工(2014年4期)2014-05-10 00:47:20
科学大众·小诺贝尔(2013年6期)2013-04-29 22:10:53
