
东亚特有植物合被韭和长梗合被韭遗传多样性的ISSR分析
2015-07-04 09:21卢利聃杨敬天周颂东何兴金
西北植物学报 2015年7期
卢利聃,杨敬天,卢 艳,周颂东,何兴金
(四川大学 生命科学学院 生物资源与生态环境教育部重点实验室,成都610064)
东亚地区是全球温带植物多样性最高的地区之一[1]。第四纪冰期(≤2.6Ma,Ma=百万年,全文统一标为Ma)时,该区域未被冰川完全覆盖,受全球气候波动影响较小,许多物种得以幸存。因此,这一区域被认为是暖温带落叶林植物在第四纪冰期气候动态变化过程中最重要的避难所,拥有大量孑遗、特有植物[2]。
特有种研究不仅在植物地理学上有极高的重要性,同时也是植物系统进化研究的关键点。以往对东亚特有种的关注常集中于一些落叶木本和松科植物上[3],较少涉及草本。但草本植物生活史短,对气候环境的变化更加敏感,因此探讨其遗传多样性及扩散机制,能使我们更好地了解东亚现存特有植物的演化历史[4]。
葱属(AlliumL.)植物长期以来作为野菜、饲料和优良牧草资源利用。其中合被组(Sect.Caloscordum)是葱属植物中一个特殊的类群,其花被片基部靠合成管状,因此称为合被。全组仅3个种,即合被韭(AlliumtubiflorumRendle)、齿棱茎合被韭(A.inutileMakino)和长梗合被韭[A.neriniflorum(Herb.)Baker.]都只分布在中国、日本,即东亚地区[5]。合被韭为中国特有,分布于中国西北及华北地区;长梗合被韭在中国华北、东北及远东地区分布。两者外部形态非常相似,并且分布区有所重叠,因而存在复杂的进化问题[6]。作为东亚特有植物的典型代表,合被韭和长梗合被韭是研究第四纪气候变化对东亚暖温带落叶林植被变迁的理想模型。但目前对其研究尚未深入,仅通过核型研究证实了两者存在紧密亲缘关系[7]。
本研究运用ISSR 分子标记[8],通过分析不同居群合被韭和长梗合被韭DNA 水平的差异和分化,揭示两个种的遗传多样性水平和遗传结构,探讨片段化生境对居群遗传多样性和遗传分化的影响,以期为解决特有种亲缘关系和物种迁移模式提供依据,从而指导制定保护策略,实现对东亚特有植物资源的保护和合理利用。
1 材料和方法
1.1 材 料
基于前人研究及标本记录,自2012年起共采集到合被韭16个居群154个个体,长梗合被韭14个居群133个个体,基本覆盖这两个种在中国整个地理分布范围(表1)。
1.2 DNA 提取和PCR 扩增
采用植物基因组DNA 试剂盒(天根生化科技有限公司,北京)提取叶片总DNA。DNA 样品用0.8%琼脂糖凝胶检测,紫外分光光度计确定纯度及浓度,稀释至40ng/L,-20℃保存。优化后体系为20μL,其中含25ng 模板DNA,1×PCR Buffer,Mg2+1.5 mmol/L,dNTPs 0.2 mmol/L,引 物0.5 μmol/L,1.0U DNATaq聚合酶。引物从UBC 第9套序列中筛选(Invitrogne公司合成),PCR 反应程序为:94 ℃5min预变性;94 ℃45s变性,特定退火温度51~57 ℃(45s)退火,72℃1 min延伸,40个循环;72 ℃10min延伸;4 ℃保存。扩增产物以Marker DL2000作为分子量标记,在含有GoldenView(Biomed基因技术有限公司,北京)的2%琼脂糖凝胶中电泳,缓冲液为1×TBE(pH 8.3)。100V恒压电泳1h,在凝胶成像系统仪(Bio-Rad Gel Doc XR+imaging system)中拍照。

表1 材料来源Table1 Sources of the material
1.3 数据处理与统计分析
电泳图谱的每条带都为一个分子标记,代表引物的一个特异结合位点。以“1”和“0”标示同一引物在相同迁移位置谱带的有无,构造数据矩阵,并记录多态性位点数(n)。采用POPGENE 1.32 计算分析遗传变异各项参数[9],包括多态位点百分率(PPB)、平均每个位点上的观察等位基因数(Na)、平均每个位点的有效等位基因(Ne)、Nei’s基因多样度指数(H)、Shannon多样性信息指数(Ho)、群体总的基因多样度(Ht)、居群内基因多样度(Hs)、居群间遗传分化系数(Gst)、基因流(Nm)和Nei’s遗传距离(D)和遗传一致度(I)。利用NTSYS-pc 2.1软件构建聚类图[10],对合被韭及长梗合被韭群体间的遗传关系进行分析。采用AMOVA 1.55分子变异分析软件,分析遗传变异在居群内及居群间的分布情况[11];遗传距离和地理距离之间的相关性运用TFPGA 1.3软件进行Mantel检验[12]。
2 结果与分析
2.1 遗传多样性水平
共筛选出谱带清晰、重复性好的6 个ISSR 引物(表2)对合被韭及长梗合被韭进行PCR 扩增,平均每个引物扩增出17条带,所得的片段长度在100~2 500bp之间。
合被韭16居群154个体共计扩增出100个清晰可重复的有效位点,其中多态位点98 个。结果(表3)显示:在物种水平上,合被韭的多态位点百分率(PPB)为98.00%,有 效 等 位 基 因(Ne)为1.419 0,Nei’s基因多样性指数(H)为0.264 8,Shannon多样性指数(Ho)为0.415 3,表明DNA 片段多态性比较丰富。各居群PPB为20.00%~64.00%,平均值为49.75%。平均有效等位基因(Ne)为1.253 4,平均Nei’s基因多样性指数(H)为0.153 1,平 均Shannon 多 样 性 指 数(Ho)为0.234 9,各参数值均低于物种水平。居群的遗传多样性综合比较表明,居群古武当山(GWDS)、怀柔(HR)各项指标大大超过平均值,遗传多样性最为丰富,而居群麦积山(MJS)、龙池曼(LCM)则相反,呈现低遗传多样性。
以同样6个引物对长梗合被韭进行PCR 扩增,共得到90个清晰可重复的有效位点,86个为多态位点。物种水平上,长梗合被韭PPB为95.56%,Ne为1.401 6,H为0.253 9,Ho为0.399 8,显示了较高的遗传多样性。居群水平上,PPB居于26.67%~63.33%,平均为48.33%;H变化范围为0.071 8~0.182 4,平均为0.145 1;Ho变化范围为0.114 0~0.280 1,平均为0.223 8,各居群内多样性均低于物种水平。其中,居群海坨山(HTS)、金阁山(JGS)、上方山(SFS)、碣石山(JSS)呈现较高遗传分化。
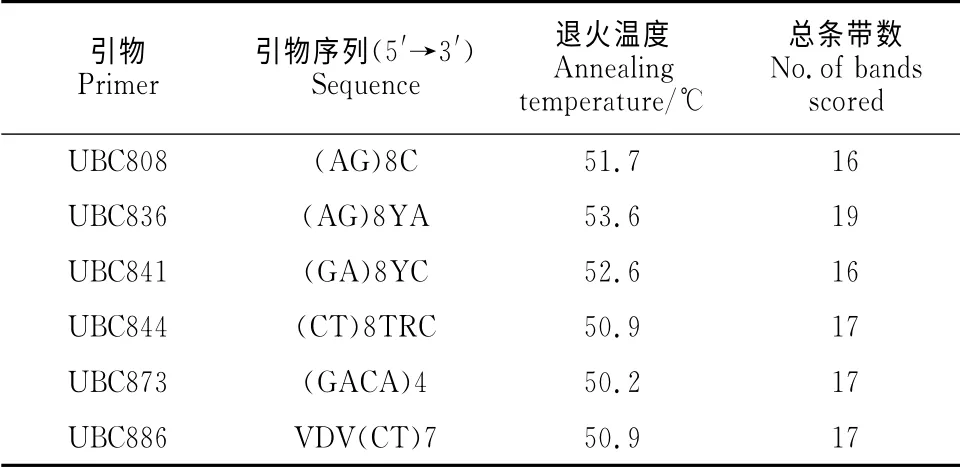
表2 ISSR 引物序列和最佳退火温度Table2 Primers and optimal annealing temperature

表3 合被韭与长梗合被韭居群的遗传多样性Table3 Genetic diversity of A.tubiflorumand A.neriniflorumpopulations
2.2 居群的遗传分化与基因流
合被韭16个居群总基因多样度(Ht)为0.264 7,居群内基因多样度(Hs)为0.153 1,Nei’s基因分化系数(Gst)为0.421 8(表4),即42.18%的变异存在于居群之间,57.82%的遗传变异存在于居群内。AMOVA 分子变异分析显示,变异率为38.95%(P<0.001),说明有38.95%的遗传变异发生在居群之间,61.05%的遗传变异存在于居群内(表5)。此结果与POPGENE分析结果一致,均表明遗传变异主要存在居群内。基因流(Nm)为0.685 4,结果反映每代居群间迁移数目小于1,居群间隔离程度较大,不足以防止遗传漂变导致的居群分化。
而长梗合被韭居群总基因多样度(Ht)为0.302 5,居群内基因多样度(Hs)为0.176 3,Nei’s基因分化系数(Gst)为0.430 1,变异率为39.17%(P<0.001),同样表明居群内的遗传分化大于居群间。基因流(Nm)为0.662 4,居群之间基因流水平也较低。
2.3 种间及居群间的遗传一致度和遗传距离分析
合被韭16个居群任意两两居群的Nei’s遗传一致度(I)范围在0.760 2~0.936 7,遗传距离(D)范围在0.065 4~0.274 2,居群间平均遗传距离为0.151 6,反映出种内遗传分化较大(表6)。长梗合被韭任意两两居群的Nei’s遗传一致度(I)范围在0.789 1~0.925 3,遗传距离(D)范围在0.077 6~0.236 9,变化趋势和合被韭基本一致(表7)。
对2 种共30 个居群进行UPGMA 聚类分析(图1),合被韭16个居群和长梗合被韭14个居群能明显分开,说明分子水平上两者出现了明显分化。但在各自种内,ISSR 标记并没有把各个居群明显的聚为几支,这反映出合被韭和长梗合被韭具有丰富的遗传变异。
Mantel检验揭示居群间地理距离和遗传距离没有 显 著 的 相 关 性(合 被 韭r=0.164 1,P=0.719 4;长梗合被韭r=0.102 7,P=0.632 2)。但从居群的地理分布上看,随纬度的变化,地理距离较近的居群聚在了一起。但在居群较为集中的区域,遗传距离与地理距离相关性较小,聚类呈现无规律状态。

表4 合被韭与长梗合被韭居群多样性Nei’s分析Table4 Nei’s analysis of gene diversity in A.tubiflorumand A.neriniflorumpopulations

表5 合被韭与长梗合被韭的AMOVA分析Table5 Analysis of molecule variance(AMOVA)of A.tubiflorumand A.neriniflorumbased on ISSR data

?

!!长梗合被"#个居群间$%&!’遗传一致度"对角线上方#和传距离"对角线下方#!"#$%&!’%(表!)*%+%(,%+-(-.韭""#/0%,("*/+"$的#"+,*%+%-(1,()-"+1%"#%$/2,("*/+"$遗#/345!"#$%&#&’()%*+6/6 7$"-(/+)群编号居8/6 7$"-(/+1/,%9:;<==<;=>!=?=!;=@=A9 BCD=DE9F ’>9;=CB9 9>=?>=9:;$GHIIJJ GHIK4K GHIIGL GHI5&I GHI5G4 GHILMI GHI5J GHI&&GHILG4 GHI4GI GHIJ54 GHI444 GH&IM4<==GH4N4K$GHIM44 GHI&&J GHIIN4 GHIIM&GHIIG5 GHI&&M GHM GHI&5L GHIJJ&GHIK&N GHIL5M GHI4J5<;=GH45M GH44JN$GHIIL4 GHI&J4 GHI555 GHILM&GHIJI5 GHII&K GHI&G4 GHI545 GHIJI GHIJKK GHMGL5>!=GH4N&J GH4LGK GH4N5L$GHIJ5J GHIJI5 GHIJJK GHI&LJ GHIKJ5 GHILML GHIL&4 GHI5JN GHI5N GHI4&5?=!;GH4KJN GH4NJJ GH4LL5 GH4J&N$GHM4K4 GHIM4L GHM4JK GHMG5N GHIM5M GHI5&M GHIKI&GHILKM GHILM4=@=GH4&5L GH44KM GH4KM4 GH4JN&GHGI&K$GHMNJL GHIMGK GHIIJK GHIJIM GHIMJL GHIJIM GHIG&4 GHIGM A9 GH4&5J GH4N&5 GH4&5&GH4JK GH44J4 GHG&&K$GHM4KI GHMGI5 GHIKI&GHI&&L GHIK4J GH&M&4 GHIGN4 BCD=GH4KIJ GH4LGN GH4JNK GH4LJN GHGIIN GH44JM GHGIKI$GHM4KL GHM GHIIJ GHI&J&GHIN&&GHIG&DE9F GH4L4L GH4GJ5 GH44MN GH455J GH4GGI GH4N4J GHGMK GHGI&5$GHMGLJ GHIII GHM45 GHI&NK GHI5JL’>GH4IKN GH4L5L GH4LM4 GH4&JN GH4444 GH4JN4 GH45GI GH4GJ5 GH4G4J$GHIK&&GHI&LJ GHI&IM GHILGI 9;=GHNGMI GH4JJI GH4&N&GH4&&I GH4KJ GH44GK GH4LGM GH4NNN GH44II GH454M$GHIKJ GHIJJM GHIGLN CB9 GH4J&&GH45NJ GH4JL4 GH4KIN GH45G&GH4JN4 GH45M GH4LNI GHGIMM GH4LJN GH45J$GHIK4K GHIJ&J 9>=GHNGM5 GH4IG5 GH4J5I GH4&4M GH4&I GHN45L GHNNK&GH4IM4 GH4LKN GH4NM GH4JJK GH45M$GHIJ&N?>=GHNLKM GHNG5 GH4G4K GHNG4&GH4&J5 GHN4N GHNNGJ GHN45J GH4KI4 GH4IJ5 GHN4MN GH4JL&GH4J54$

图1 合被韭和长梗合被韭居群的UPGMA 聚类图Fig.1 UPGMA dendrogram of A.tubiflorumand A.neriniflorum
3 讨 论
3.1 遗传多样性水平分析
本研究中合被韭及长梗合被韭的PPB均达到95.00%以上,明显高于Nybom 和Bartish 总结的物 种普遍的平均遗传多样性PPB值(71.02%)[13]。同时,物种水平的基因多样性指数均高于单子叶植物的平均值0.19[14],说明两者在物种水平上都具有很高的遗传多样性。
新种形成[15]、繁育系统[16]、多次奠基者效应[17]、冰期残余种[18]等诸多因素,都有可能导致区域特有物种具有高水平遗传多样性。葱属是一起源相对古老的类群[19],遗传变异丰富,相当于被子植物其他类群的科或亚科级水平[20]。合被组所在的葱属第二进化支,起源于始新世晚期(约34.93 Ma)。渐新世晚期和中新世早期(约21.23 Ma),合被组祖先类群开始分化。此时青藏高原隆升到相当高度,东亚季风气候形成,中国北方出现强烈的干旱,一直延伸到中国东部的沿海地带[19,21]。在野外调查中,我们观察到合被韭具有二次生长的特性,正是其对恶劣环境的适应性进化。因此,合被韭及长梗合被韭种级水平上很高的遗传多样性,可能与其复杂的遗传背景及生境有关。
但在居群水平上,两者的平均多态位点百分率低于50.00%,均低于物种水平,表明两者居群内部的遗传多样性明显不足。这种现象的成因可能是由于合被韭与长梗合被韭具有以鳞茎分裂无性繁殖的生活史特性;也可能是由于近年中国北方干旱气候不断加剧,植被破坏严重,导致生境片段化,从而造成的野生居群间基因流受阻。
3.2 遗传结构与进化机制
基于POPGENE和AMOVA 统计分析均显示两个种的遗传变异主要存在于居群内。两者遗传分化系数Gst均高于Nybom 统计的植物ISSR 遗传分化系数的平均值0.34[22],表明其已形成异质居群结构。
前人研究表明长梗合被韭中存在杂合现象,随体呈杂合状态,同一个细胞既有末端随体,又有居间随体[7]。同时,该研究显示,合被韭与长梗合被韭核型基本一致,普遍为二倍体,但合被韭北京的一个居群为四倍体,表明该类群在扩大生存空间过程中不仅出现了染色体结构变异,还出现了染色体加倍[7]。普遍认为杂合优势和多倍化是葱属植物进化的主要驱 动力[23-24]。因此,结合本研究ISSR遗传多样性和前人的核型资料,认为合被韭和长梗合被韭的遗传结构变异和遗传分化,是通过杂交和染色体加倍的方式实现的。通常认为,异交物种的遗传变异主要分布在居群内[25],而杂交和染色体加倍过程中会使得育性降低、居群缩小。但葱属植物中绝大多数物种均利用鳞茎、根状茎或珠芽等繁育方式[24],来弥补多倍化和杂交的不利作用。合被韭和长梗合被韭均具鳞茎,且野外考察中发现这两个种往往多株成丛生长。因此认为合被韭和长梗合被韭的进化驱动力是杂交和多倍化,兼行无性繁殖则可克服育性降低带来的居群个体数量下降,保存其变异。合被韭与长梗合被韭复杂的遗传背景和多样化的繁育方式极可能是导致居群内遗传分化显著的重要因素。
基因流一直被视为是使居群遗传结构均质化的主要因素之一[26]。当基因流小于1时,就不足以抵制居群内因遗传漂变而引起的遗传分化[27]。本研究中,两种东亚特有植物的基因流Nm均小于1,远低于Hamrick 总结的一般广布种植物的基因流(Nm,1.881)[28],因此种群隔离引起的遗传漂变也是影响其遗传分化的重要因素。
3.3 合被韭和长梗合被韭的分化中心和遗传多样性中心
对居群的遗传多样性综合指标进行比较,结果表明,燕山山脉、太行山脉及邻近地区的合被韭(居群GWDS、HR)遗传多样性最为丰富,同一区域的长梗合被韭(居群HTS、JGS、SFS、JSS)也同样呈现较高的遗传分化。这一地区不仅是合被韭和长梗合被韭遗传多样性最丰富的地区,也恰好是两者分布的重叠区。UPGMA 聚类显示,合被韭在这一区域(居群GWDS、HR)亲缘关系接近,长梗合被韭在该区域(居群HTS、JGS、SFS、JSS),以及邻近的环渤海湾区域(居群CD、NH)也表现出很紧密的亲缘关系。根据葱属系统发育研究资料[21],合被韭与长梗合被韭在系统发育树中始终聚为一个独立的单系分支。从种级水平上看,此单系分支中这两个种互为姊妹类群。本研究UPGMA 聚类也支持两者为单系,说明合被韭与长梗合被韭应拥有一个最近的共同祖先类群,且这个祖先类群的分布区域应是合被韭和长梗合被韭的分布重叠区域。2个种丰富的遗传变异,暗示其祖先类群亦具丰富的遗传基础。因此,燕山山脉、太行山脉及邻近地区可能是合被韭及长梗合被韭的分化中心和遗传变异中心。
遗传多样性是植物对遗传基础和生境的响应,显示高水平遗传多样性的地区,通常暗示着该区域历史上曾是物种的冰期避难所[29]。气候波动时,冰期避难所相对稳定的生态环境和气候,能促使生长在该区域的物种积累丰富的遗传多样性[30]。在野外调查中发现合被韭具有休眠,或者说二次生长的特性。这暗示其进化史上可能经历了生态环境大的改变,比如干旱、低温等,迫使其不得不进入休眠状态,在严苛的生境中保存繁殖能力。古植被的重建显示,中国东部-日本南部-朝鲜半岛曾被一条长的温带落叶林覆盖[1]。古孢粉学证据也显示,中国东部海岸被淹没的大陆架存在温带落叶林的冰期避难所[2]。目前虽然并没有研究证实燕山山脉及太行山脉地区曾存在冰期避难所,但由于该地区地貌在燕山期(约199.6~133.9 Ma)已经形成,受喜马拉雅造山运动及第四纪冰川作用的影响相对较弱,在气候波动下,可能会有较适宜植物存活的温度。
合被韭与长梗合被韭的外部形态非常相似[6],说明祖先性状得到了充分保留。进化史上相似的遗传背景,则使得两者进化出了相似的遗传适应策略,在适宜生存的地区保留更高的遗传多样性,没有表现出显著的表型分化。而因为山脉、沟谷等小地形的差异,表现出居群内遗传多样性的强烈变化。
目前,合被组仅有的3个种中,本研究尚未涉及齿棱茎合被韭(A.inutile)也只分布于东亚。但该种在中国鲜有报道,仅有1条资料显示在安徽滁县琅琊山有分布[31],其余记录均来自于日本。笔者及实验室研究团队曾在3年间(2012~2014)的盛花期多次赴安徽滁县,但未见其踪影。推测合被组祖先类群在扩张的过程中,曾产生了一些亲缘关系非常近的类群,但受第四纪地质气候变化的影响及生境片断化的不断加剧,这些类群经历了严重的灭绝和重扩张过程。其中一些类群已经绝灭,而齿棱茎合被韭原有分布区逐步坍缩,形成岛屿间断分布格局。
因此,虽然物种水平上,合被韭和长梗合被韭尚保留其祖先丰富的总体遗传多样性,但在居群内其遗传基础已逐渐趋于一致,并且居群间基因流有限,这使其不能有效地维持目前的遗传多样性水平。高遗传多样性的物种,如果不能有效维持居群大小和自然更新,同样可能引起居群的退化甚至灭绝[32]。
居群衰退是特有植物普遍存在的威胁。东亚地区拥有大量孑遗、特有植物,更应高度重视气候和环境变化对这一区域植物遗传多样性的影响,维持这一地区的稳态。为保持丰富的物种遗传多样性水平,维持生物多样性,应尽可能避免人为加速生境的片段化。这对于降低物种濒危机率,保护特有类群,实现对东亚特有植物资源的保护和合理利用有着长远的意义。
[1] HARRISON S P,YU G,TAKAHARA H,etal.Diversity of temperate plants in East Asia[J].Nature,2001,413(6 852):129-130.
[2] QIU Y X,FU C X,COMES H P.Plant molecular phylogeography in China and adjacent regions:Tracing the genetic imprints of Quaternary climate and environmental change in the world's most diverse temperate fiora[J].MolecularPhylogeneticsandEvolution,2011,59:225-244.
[3] MENG H H,ZHANG M L.Phylogeography ofLagochilusilicifolius(Lamiaceae)in relation to Quaternary climatic oscillation and aridification in Northern China[J].BiochemicalSystematicsEcology,2011,39(4-6):787-796.
[4] ZHAO C,WANG C B,MA X G,etal.Phylogeographic analysis of a temperate-deciduous forest restricted plant(BupleurumlongiradiatumTurcz.)reveals two refuge areas in China with subsequent refugial isolation promoting speciation[J].MolecularPhylogenetics andEvolution,2013,68(3):628-643.
[5] HANELT P,SCHULZE M J,FRITSCH R M,etal.Infrageneric Grouping ofAllium—the Gatersleben Approach[M]//HANELT P,HAMMER K,KNUPFFER H,etal.The GenusAllium:Taxonomic Problems and Genetic Resources.Germany:Gatersleben,1992:107-123.
[6] XU J M,RUDOLF V,KAMELIN.Flora of China[M].Beijing:Science Press,2000,24:201-202.
[7] LI R J(李汝娟),SHANG Z Y(尚宗燕),CUI T CH(崔铁成),etal.Studies on karyotypes and phylogenetic relationship ofAlliumsect.Caloscordum(Liliaceae)from China[J].ActaPhytotaxonomicaSinica(植物分类学报),1996,34(3):288-295(in Chinese).
[8] ZIETKIEWICE E,RAFALAKE A,LABUDA D.Genome fingerprinting by simple sequence repeat(SSR)anchored polymerase chain reaction amplification[J].Genomics,1994,20(2):176-183.
[9] YEH F C,YANG R C,BOTLE T B J,etal.POPGENE:The User Friendly Shareware for Population Genetic Analysis(Ver.1.32)[M].Canada:Molecular Biology and Biotechnology Centre,University of Alberta,1997.(program available from:http://www.ualberta.ca/~fyeh/)
[10] ROHLF F J.NTSYS-pc:Numerical Taxonomy and Multivariate Analysis System(Ver.2.1)Exeter Software[CP/DK].New York:Setauket,1998.
[11] EXCOFFIER L.AMOVA:Analysis of Molecular Variance(Ver.1.55)[CP/DK].Switzerland:Genetics and Biometry Laboratory,University of Geneva,1995.
[12] MILLER M P.TFPGA:Tools for Population Genetic Analysis(Ver.1.3)[Z].Arizona:Department of Biological Sciences,Northern Arizona University,1997.
[13] NYBOM H,BARTISH I V.Effects of life history traits and sampling strategies on genetic diversity estimates obtained with RAPD markers in plants[J].PerspectivesinPlantEcologyEvolutionandSystematics,2000,3(2):93-114.
[14] LI Q,XIAO M,GUO L,etal.Genetic diversity of the rare and endangered plantTrilliumtschonoskiiin Sichuan Province[J].Journalof BeijingForestryUniversity,2005,27(4):1-6.
[15] RANKER T A.Evolution of high genetic variability in the rare Hawaiian fernAdenophorusperiensand implications for conservation management[J].BiologicalConservation,1994,70(1):19-24.
[16] LEWIS P O,CRAWFORD D J,LEWIS P O.Pleistocene refugium endemics exhibit greater allozymic diversity than widespread congeners in the genusPolygonella(Polygonaceae)[J].AmericanJournalofBotany,1995,82(2):141-149.
[17] MAGUIRE T L,SEDGLEY M.Genetic diversity inBanksiaandDryandra(Proteaceae)with emphasis onBanksiacuneata,a rare and endangered species[J].Heredity,1997,79(1):394-401.
[18] ZAWKO G,KRAUSS S L,DIXON K W,etal.Conservation genetics of the rare and endangeredLeucopogonobtectus(Ericaceae)[J].MolecularEcology,2001,10(10):2 389-2 396.
[19] FRIESEN N,FRITSCH R M,POLLNER S,etal.Molecular and morphological evidence for an origin of the aberrant genusMilulawithin Himalayan species ofAllium(Alliaceae)[J].MolecularPhylogeneticsandEvolution,2000,17(2):209-218.
[20] BALDWIN B G,DONOGHUE M J.The ITS region of nuclear ribosomal DNA avaluable source of evidence on angiosperm phylogeny[J].AnnalsoftheMissouriBotanicalGarden,1995,82(2):247-277.
[21] LI Q Q,ZHOU S D,HE X J,etal.Phylogeny and biogeography ofAllium(Amaryllidaceae:Allieae)based on nuclear ribosomal internal transcribed spacer and chloroplastrps16sequences,focusing on the inclusion of species endemic to China[J].AnnalsofBotany,2010,106(5):709-733.
[22] NYBOM H.Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants[J].MolecularEcology,2004,13(5):1 143-1 155.
[23] HOGBIN P M,PEAKALL R.Evaluation of the contribution of genetic research to the management of the endangered plantZieriaprostrate[J].ConservationBiology,1999,13(3):514-522.
[24] ZHOU S D(周颂东),HE X J(何兴金),YU Y(余 岩),etal.Karyotype studies on twenty-one populations of eight species inAlliumsectionRhiziridium[J].ActaPhytotaxonomicaSinica(植物分类学报),2007,45(2):207-216(in Chinese).
[25] ZHOU CH J(周春景),ZHOU S D(周颂东),HUANG D Q(黄德青),etal.Karyotypes of 25populations of 15species inAlliumSectionRhiziridiumfrom China[J].PlantDiversityandResources(植物分类与资源学报),2012,34(2):120-136(in Chinese).
[26] VEKEMANS X,HARDY O J.New insights from fine-scale spatial genetic structure analyses in plant populations[J].MolecularEcology,2004,13(4):921-935.
[27] SLARKIN M.Gene flow in natural populations[J].AnnualReviewofEcologyandSystematics,2003,16:393-430.
[28] HAMRICK J L.Gene Flow and Distribution of Genetic Variation in Plant Populations[M].New York:Academes Press,1987,53-67.
[29] TZEDAKIS P C,LAWSON I T,FROGLEY M R,etal.Buffered tree population changes in a Quaternary refugium:evolutionary implication[J].Science,2002,297(5 589):2 044-2 047.
[30] GONG W,CHEN C,DOBEšC,etal.Phylogeography of a living fossil:Pleistocene glaciations forcedGinkgobilobaL.(Ginkgoaceae)into two refuge areas in China with limited subsequent postglacial expansion[J].MolecularPhylogeneticsandEvolution,2008,48(3):1 094-1 105.
[31] WANG X H(王希华),QIAN SH X(钱士心),WAN ZH G(万志刚).Alliuminutile,a new record species to China[J].ActaBotanica Yunnanica(云南植物研究),1999,21(4):1-3(in Chinese).
[32] LI A(李 昂),GE S(葛 颂).Advances in plant conservation genetics[J].BiodiversityScience(生物多样性),2002,10(1):61-71(in Chinese).
猜你喜欢
河北科技师范学院学报(2022年2期)2022-08-26
浙江中医药大学学报(2021年6期)2021-07-12
生态学报(2021年3期)2021-03-31
中国粮油学报(2020年12期)2021-01-09
海南医学(2020年1期)2020-01-18
绿色科技(2019年14期)2019-11-19
江苏农业科学(2019年5期)2019-09-02
电子制作(2019年24期)2019-02-23
广东农业科学(2017年5期)2017-08-29
中国康复理论与实践(2015年10期)2015-12-24
