
密度泛函理论计算姜黄素结合Pb(Ⅱ)
2015-06-24 14:28何永辉李志江彭嘉鲜郭俊明
云南民族大学学报(自然科学版) 2015年2期
何永辉,田 宇,李志江,彭嘉鲜,郭俊明
(云南民族大学 民族药资源化学国家民委-教育部重点实验室,云南 昆明 650500)
密度泛函理论计算姜黄素结合Pb(Ⅱ)
何永辉,田 宇,李志江,彭嘉鲜,郭俊明
(云南民族大学 民族药资源化学国家民委-教育部重点实验室,云南 昆明 650500)
采用纯密度泛函BP86和杂化泛函B3LYP计算了姜黄素结合Pb(Ⅱ)的结构,并分析其成键性质.计算结果显示,2个姜黄素分子结合1个Pb(Ⅱ)成Pb-L2结构具有更低的能量,首次发现Pb(Ⅱ)结合4个O原子形成Pb-O4的半球形(hemisphere)配位结构,其中2个Pb-O键长较短(0.220 nm),另外2个Pb-O键长则较长(0.236 nm).进一步分析Pb-O键的性质,指出6S2孤对电子未参与成键,因而具有立体化学活性.这为进一步研究姜黄素的药理作用提供了理论基础.
密度泛函理论;姜黄素;铅
傣药双姜胃痛丸由姜黄、紫色姜、地不容、石菖蒲、苦菜子、蜂蜜( 炼) 等药材组成,具有理气止痛、和胃降逆的功效,主要用于中焦气滞所致的胃满胀痛、嗳气吞酸、慢性浅表性胃炎等[1].姜黄素是双姜胃痛丸中的主要成份,具有杀菌、抗炎、抗肿瘤,以及治疗老年痴呆症等生理活性[2].姜黄素作为一种金属离子络合剂,阻止β淀粉样蛋白(beta-Amyloid,Aβ)的聚积作用已经为人们所接受[3].
铅污染是影响未成人年生长发育的一个严重环境问题,解铅毒性的相关研究一直是科学研究的热点.姜黄素作为一种强的金属离子络合剂,其与Pb(Ⅱ)结合的研究较少见诸报导.姜黄素分子结构有酮式和烯醇式2种,在水溶液中以烯醇式结构存在时,具有更低的能量.同时,由于烯酮式结构存一个二齿配体,易与金属离子结合[4].但目前尚不清楚姜黄素与Pb(Ⅱ)结合的配位结构、成键性质等相关参数.
采用密度泛函理论计算姜黄素结合Pb(Ⅱ)的自由能变化,从理论上确定其配位结构为Pb-(curcumin)2型,即1个Pb(Ⅱ)结合4个O形成Pb-O4的半球形(hemisphere)配位结构,并进一步分析了Pb-O键的性质.
1 计算部分
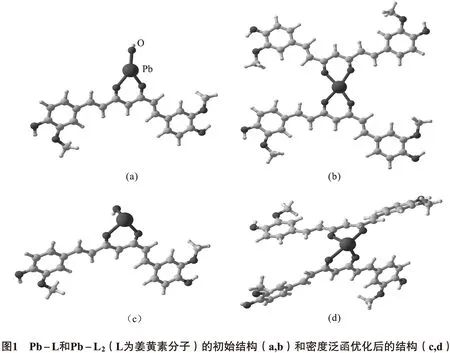
完成所有计算的硬件为Intel 16核46 GB内存,量子化学计算软件为Gamess[5],分子构建软件为MacMolPlt(版本为7.4.4).姜黄素与Pb(Ⅱ)结合的主要可能结构有:Pb(Ⅱ)与1个姜黄素分子中2个O和1个羟基O结合1个形成三配位结构Pb-O3,或是Pb(Ⅱ)与2个姜黄素分子中的O结合形成四配位结构Pb-O4.这2种结构的初始结构模型如图1(a,b)所示,其分子点群为C1.利用基于纯密度泛函理论的BP86和杂化泛函B3LYP对体系进行计算.基组的选择为:O、C、H采用aug-cc-pVTZ基组,Pb则采用包含赝势的aug-cc-pVTZ-PP基组[6].在没有对称性限制条件下,完成结构优化,然后采用相同的理论计算水平计算频率,确定所得结构均为势能面上的稳定点.考虑分子位于水溶液环境中,采用导体极化连续模型(CPCM)考虑溶剂效应[7].
2 结果与讨论
2.1 分子结构
由于姜黄素在溶液中主要以烯醇式结构存在,易与Pb(Ⅱ)形成Pb-O键的配位结构.同时,姜黄素分子对配位结构有较大的空间位阻,不易与Pb(Ⅱ)形成高配位数的配位结构.据此,计算其中最有可能形成的两种配位结构: Pb-L和Pb-L2(L表示配体姜黄素).初始结构采用所有配体共平面的结构,在没有对称性限制的条件下,采用2种泛函BP86和B3LYP计算2种配位结构的优化结构,初始结构及优化结构如图1所示.从图中可见,2种优化结构Pb-L和Pb-L2的配体都位于Pb(Ⅱ)配位平面之下, Pb(Ⅱ)的孤对电子占据轴向的位置,表明其孤对电子有明显的立体化学活性.这个计算结果与文献的相关报道一致,即Pb(Ⅱ)在低配位数结构中,其孤对电子具有立体化学活性[8].
化合物的稳定性可以通过计算对应的结合能进行判断.在考虑溶剂效应、零点能和热力学校正的情况下,分别计算反应物和产物的自由能和熵值,即可得到化合物的结合能.通过计算获得了姜黄素分子、Pb(Ⅱ)、羟基和形成的2种产物(Pb-L和Pb-L2)在水溶液环境中的吉布斯自由能值和熵值,并计算了生成Pb-L和Pb-L2反应的自布斯自由能变化值,结果列于表1.从表中可知,2种泛函都预测模型化合物Pb-L2具有更低的结合能,表明该化合物在热力学上更优先.
注:a基于密度泛函计算得到的能量值(包含溶剂效应和零点能的校正).
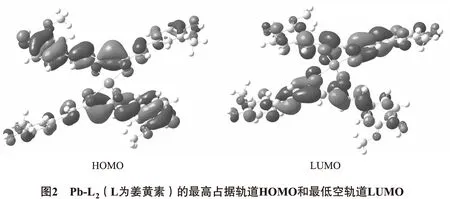
在Pb-L2结构中,Pb(Ⅱ)与2分子姜黄素的4个氧原子形成Pb-O4的四配位结构,其中2个键位于短对角线,Pb-O键长较短(0.220 nm),另外2个键则位于另一条较长的对角线,Pb-O键长为0.236 nm.该较长的键(0.236 nm)为羰基氧与Pb(Ⅱ)成键,说明该Pb-O键的成键能力较弱.文献报道,在形成包含Pb-O键的四配位结构中,其中Pb-O键会变长,甚至达到0.240 nm[9].这是由于铅的孤对电子对配体的排斥作用所导致.此外,姜黄素与Pb形成Pb-L2结构,2个姜黄素配体平面夹角约90°,更加有利于与Aβ中的疏水结构形成π-π堆积,阻止Aβ单体的聚积.相关的文献报道也证实,姜黄素分子结合金属离子后,具有更强的抗Aβ聚积作用[10].从前线分子轨道图中也可以看出(图2),2分子姜黄素与Pb(Ⅱ)结合形成Pb-L2后,其最高占据轨道(highest occupied molecular orbital,HOMO)由姜黄素分子的大π键组成,最低空轨道(lowest unoccupied molecular orbital,LUMO)为姜黄素分子的大反π键(π*键)组成.因此,Pb-L2能与Aβ单体形成π-π堆积.
2.2 成键分析
为了进一步理解化合物Pb-L2的结构性质,我们采用杂化泛函B3LYP计算了其中Pb-O键的性质.首次分析了化合物Pb-L2的电子分布,计算结果如表2所示.结果表明,醇氧所带电荷随配体O数目的增大而增大,这是因为来自于Pb(Ⅱ)的平衡正电荷一定.同时,发现Pb(Ⅱ)的有效电荷与配体个数相关,随配体个数的增大而减小.

表2 Pb-L2与Pb(Ⅱ)结合的配体O的电子布局
注:O(1)为醇氧,O(2)为羰基上的氧.
表3列出了化合物Pb-Ln(n=1~2)的分子轨道性质.Pb的原子轨道在Pb-O键中的比重随着配体数的增加而增加,从22.80%增大至29.32%.相应的Pb-O键键长也随着配体数的增加而增长.在Pb(Ⅱ)的成键轨道中,Pb-O键主要由Pb的6P轨道组成,而Pb(Ⅱ)的6S轨道几乎没有参与成键,即6S轨道明显为惰性轨道.因此,Pb(Ⅱ)的6S2电子具有立体化学活性.

表3 化合物Pb-L2的成键分析
注:O(1)为醇氧,O(2)为羰基上的氧.
3 结语
采用密度泛函BP86和B3LYP计算了姜黄素结合Pb(Ⅱ)的结构及成键性质.结果表明, Pb(Ⅱ)结合2分子姜黄素形成Pb-L2结构具有更低的能量,首次发现Pb(Ⅱ)结合4个O原子形成Pb-O4的半球形(hemisphere)配位结构,其中2个Pb-O键长较短(0.220 nm),另外2个Pb-O键长则较长(0.236 nm).并分析了Pb-L2结构中Pb-O键的性质,为进一步研究姜黄素的药理作用提供了理论基础.
[1] 李维,王剑,杜西铃,等.HPLC法测定傣药双姜胃痛丸中姜黄素的含量[J].中国民族医药杂志,2008,13(10):53-55.
[2] 韩婷,宓鹤鸣.姜黄的化学成分及药理活性研究进展[J].解放军药学学报,2001,17(2):95-97.
[3] PICCIANO A L,VADEN T D.Complexation between Cu(Ⅱ)and curcumin in the presence of two different segments of amyloid β[J].Biophys Chem,2013,184:62-67.
[4] KOLEV T M,VELCHEVA E A,STAMBOLIYSKA B A,et al.DFT and experimental studies of the structure and vibrational spectra of curcumin[J].J Quantum Chem,2005,102(6):1069-1079.
[5] SCHMIDT M W,BALDRIDGE K K,BOATZ J A,et al.General atomic and molecular electronic structure system[J].J Comput Chem,1993,14(11):1347-1363.
[6] PETERSON K A.Systematically convergent basis sets with relativistic pseudopotentials.I.Correlation consistent basis sets for the post-d group 13-15 elements[J].J Chem Phys,2003,119(21):11009-11112.
[7] BARONE V,COSSI M.Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model[J].J Phys Chem A,1998,102(11):1995-2001.
[8] DAVIDOVICH R L,STAVILA V,MARININ D V,et al.Stereochemistry of lead(Ⅱ)complexes with oxygen donor ligands[J].Coord Chem Rev,2009,253(9/10):1316-1352.
[9] HE Y,LIU M,DARABEDIAN N,et al.pH-dependent coordination of Pb2+to metallothionein 2:structures and insight into lead detoxification[J].Inorg Chem,2014,53(6):2822-2830.
[10] PALLIKKAVIL R,UMMATHUR M B,SREEDHARAN S,et al.Synthesis,characterization and antimicrobial studies of Cd(Ⅱ),Hg(Ⅱ),Pb(Ⅱ),Sn(Ⅱ)and Ca(Ⅱ)complexes of curcumin[J].Main Group Met Chem,2013,36(3/4):123-127.
(责任编辑 梁志茂)
Density functional calculation of curcumin binding to Pb(Ⅱ)
HE Yong-hui,TIAN Yu,LI Zhi-jiang,PENG Jia-xian,GUO Jun-ming
(Key Laboratory of Chemistry in Ethnic Medicinal Resources,State Ethnic Affairs Commission and Ministry of Education of China,Yunnan Minzu University,Kunming 650031,China)
Two possible structures of curcumin binding to Pb(Ⅱ) have been calculated by the density functional(pure functional BP86 and hybrid functional B3LYP),and the Pb-O bond characterization has also been detected. It is demonstrated that the coordination structure Pb-L2(L refers to curcumin) is preferable thermodynamically,and the structure mode Pb-O4with the hemisphere coordination has been found for the first time. In the Pb-O4structure,there are two short Pb-O bonds(0. 220 nm),and two Pb-O bonds with a longer distance(0.236 nm). The further analysis of the characterization of Pb-O shows that the 6S2lone pairs are active stereochemically. This work will be helpful for further study on the pharmacological effects of curcumin.
density functional theory; curcumin; lead
2014-11-25.
国家自然科学基金(51262031);云南省应用基础研究项目(2013Y022);云南民族大学化学与生物技术学院SRT项目(2013HXSRT01);民族药资源化学重点实验室开放课题(MZY1402);云南民族大学青年基金(2013QN21).
何永辉(1981-),男,博士研究生.主要研究方向:生物无机化学.
郭俊明(1962-),男,教授,硕士生导师.主要研究方向:锂离子电池正极材料制备与性能研究.
O614.43
A
1672-8513(2015)02-0147-04
猜你喜欢
天津农学院学报(2022年2期)2022-08-05
渤海大学学报(自然科学版)(2022年1期)2022-07-25
九江学院学报(自然科学版)(2022年2期)2022-07-02
云南大学学报(自然科学版)(2022年3期)2022-05-25
包装与食品机械(2021年5期)2021-11-06
华南师范大学学报(自然科学版)(2021年1期)2021-03-09
家庭百事通·健康一点通(2021年1期)2021-02-24
当代陕西(2019年6期)2019-04-17
文萃报·周五版(2018年25期)2018-08-13
分析化学(2018年2期)2018-03-02
