
小反刍兽疫病毒N蛋白B细胞表位预测、合成及间接ELISA方法的建立
2015-06-18 11:28曾少灵阮周曦唐金明廖立珊林庆燕吕建强叶奕优李希强王红英杨俊兴花群义
动物医学进展 2015年8期
曾少灵,阮周曦,唐金明,廖立珊,孙 洁,林庆燕,吕建强,叶奕优,祝 贺,李希强,王红英,杨俊兴,花群义*
(1.深圳出入境检验检疫局动植物检验检疫技术中心,广东深圳 518045;2.云南出入境检验检疫局检验检疫技术中心,云南昆明 650028;3.内蒙古牙克石市动物疫病防控中心,内蒙古牙克石 022150)
小反刍兽疫(Peste des petits ruminants,PPR)是由小反刍兽疫病毒 (Peste des petits ruminants virus,PPRV)引起的一种急性传染病,主要感染绵羊和山羊,其中山羊高度易感,野生小反刍兽偶有发生[1-2]。此病以发热、口炎、腹泻、肺炎为特征[3],严重暴发时病死率可高达100%[2,4]。世界动物卫生组织(OIE)将其列为必须报告的动物疫病,我国将其列为一类动物疫病[2-3]。此病2007年首次传入我国,在西藏地区发生并一直流行,之后由边境逐渐向内地传播[1,5],至2014年4月大范围暴发,已波及超过17个省市地区[3,5]。目前,PPR已成为威胁我国养羊业发展的重要疫病,造成大批羊只死亡,经济损失巨大[3,6]。
PPRV共编码6种结构蛋白和2种非结构蛋白,其中N蛋白是PPRV结构蛋白中的核衣壳蛋白,具有含量最丰富、免疫原性最强和特异性最好等特点,在病毒感染的动物血清中针对N蛋白的抗体占主导地位[7-10]。本研究通过生物软件分析获取PPRV N蛋白的B细胞抗原表位信息,据此合成了含有特异性抗原表位的人工多肽,并用作包被抗原,建立了PPRV血清抗体间接ELISA方法,对于该病的研究和预防控制具有重要意义。
1 材料与方法
1.1 材料
1.1.1 人工合成多肽 多肽片段由深圳出入境检验检疫局动植物检验检疫技术中心动检室设计,由上海强耀生物科技有限公司负责合成。
1.1.2 试剂和仪器 山羊小反刍兽疫标准阳性血清和标准阴性血清、山羊牛瘟病毒免疫阳性血清从英国动物卫生研究所Pirbright实验室引进;50份疫区临床山羊阳性血清、80份非疫区山羊阴性血清由西藏出入境检验检疫局技术中心提供;山羊口蹄疫病毒阳性血清、山羊痘病毒阳性血清、山羊棘球蚴阳性血清均为深圳出入境检验检疫局动植物检验检疫技术中心动检室保存;法国小反刍兽疫标准竞争ELISA检测试剂盒(CIRAD-BIOS);兔抗山羊IgGHRP、底物显色液等试剂为Invitrogen公司产品;I MARK型自动酶标仪;Eppendorf 5415R型高速离心机;CTC-256型恒温恒湿培养箱等。
1.2 方法
1.2.1 蛋白序列的获取 根据序列号AAS68026.1从GenBank下载获得PPRV病毒N蛋白氨基酸全序列。
1.2.2 二级结构的分析 综合采用Gamier-Robson、 Chou-Fasman、 Kyte-Doolittle、 Esenberg、Karplus-Schulz、Jameson-wolf和 Emini 7种预测方法分析PPRV病毒N蛋白的二级结构[11-12]。
1.2.3 B细胞表位的预测 在1.2.2二级结构分析结果基础上,推断PPRV病毒N蛋白可能的B细胞表位[11-12]。
1.2.4 多肽的设计和人工合成 基于1.2.3的B细胞表位推断结果,设计几段多肽,交付多肽合成上海强耀生物科技有限公司进行人工合成。
1.2.5 利用人工合成多肽建立间接ELISA检测方法[11,13-16]用pH 9.2的碳酸盐缓冲液将人工合成的多肽稀释成一定浓度。在酶标板的每孔内加入100μL多肽包被液,于4℃冰箱中包被16h~24h。以PBST洗液清洗3次。每孔加入50g/L的明胶液100μL,37℃下封闭2h。以PBST洗液洗涤3次,备用。分别对小反刍兽疫标准阳性血清和标准阴性血清按1∶25、1∶50、1∶100、1∶200进行稀释,与小反刍兽疫疫区临床山羊阳性血清、非疫区山羊阴性血清,以及山羊牛瘟病毒超免疫阳性血清、山羊口蹄疫病毒阳性血清、山羊痘病毒阳性血清和山羊棘球蚴阳性血清一起作为待测样品,采用包被好的ELISA板进行间接ELISA(iELISA)检测。加入各种血清样品后,37℃下孵育45min。以PBST洗液洗涤3次。按100μL/孔加入1∶5 000稀释的兔抗山羊-IgG,37℃下孵育30min。显色,终止反应,测定OD 450nm的值。使用检测后确认特异性和敏感性良好的人工多肽包被ELISA板,分批存放在-20℃和4℃,间隔不同时间段,取出用于检测,测试包被物的稳定性。
1.2.6 临床血清样本的检测和与进口试剂盒的符合率试验 用1.2.5建立的间接ELISA方法和进口的标准小反刍兽疫血清抗体检测试剂盒同时对小反刍兽疫疫区临床山羊阳性血清和非疫区山羊阴性血清进行检测,统计两种方法的检测符合率。
2 结果
2.1 PPRV N蛋白序列的获取与初步分析
从GenBank下载获得PPRV N蛋白氨基酸全序列,序列号为AAS68026.1。利用DNA Star软件对此序列进行初步分析,推断N蛋白的B细胞抗原表位可能集中在靠近C-端的由389个氨基酸组成的肽段中,长度约为全蛋白序列的2/3[8,12]。本文作为研究目标的蛋白氨基酸序列如图1。
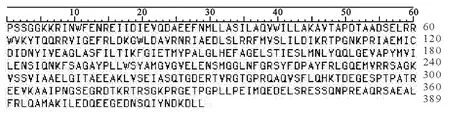
图1 作为本文研究目标的PPRV N蛋白氨基酸序列Fig.1 The amino acid sequences of N protein in PPRV as a research target in this study
2.2 PPRV N蛋白的二级结构分析结果
以Gamier-Robson、Chou-Fasman、Kyte-Doolittle和Esenberg等分析方法,预测PPRV N蛋白α-螺旋、β-折叠、转角、无规则卷曲等结构,以及亲水高峰区、两亲性区域、柔性区域、抗原性指数区域和表面可及性区域等二级结构和特殊区域的位置,结果如图2所示。当两种分析方法对同一结构存在的区域预测结果不一致时,优先采用两种方法均确认的区域作为该种结构的预测位置。以峰形图展示的结构分析,均采用峰值大于基线值(0或1)的区域对应的预测位置。
2.3 PPRV N蛋白的B细胞表位预测结果
综合2.2的分析结果,选取PPRV N蛋白氨基酸区段中没有α-螺旋或β-折叠结构,满足抗原性指数≥0、亲水性指数≥0和氨基酸的表面可及性指数≥1等条件的区段,在上述区段的内部或附近再寻找具有转角结构、无规则卷曲和柔性结构的区段,最后推断出N蛋白的抗原表位可能处在蛋白N端77-79、108-111、187-190、191-192、212-213、218-222、223-226、238-240、289-292、294-296、313-316、317-327和384-386位置的氨基酸区段内或附近。
2.4 PPRV N蛋白含B细胞抗原预测表位的人工多肽的设计和合成
在PPRV N蛋白中选取了4个区段,每个区段长度约为50个~60个氨基酸,分别包含2.3预测分析得出的B细胞抗原表位,交付专业公司完成人工多肽的合成。选取的4段人工合成多肽的氨基酸序列及编号分别为:①61-120位氨基酸区段,编号为PPRVN(61-120),氨基酸序列为 WVKYTQQRRVIGEFRLDKGWLDAVRNRIAEDLSLRRFMVSLILDIKRT PGNKPRIAEMIC;②185-244位氨基酸区段,编号为PPRV-N(185-244),氨基酸序列为IQNKFSAGAYP LLWSYAMGVGVELENSMGGLNFGRSYFDPAY FRLGQEMVRRSAGKVSSV;③264-323位氨基酸区段,编号为 PPRV-N(264-323),氨基酸序列为SQTGDERTVRGTGPRQAQVSFLQHKTDEGES PTPATRE EVKAAIPNGSEGRDTKRTRSGK;④306-365位氨基酸区段,编号为PPRV-N(306-365),氨基酸序列为AIPNGSEGRDTKRTRSGKPR GETPGPLLPEIMQEDELSRESSQNPREAQRSA EALRLQA。
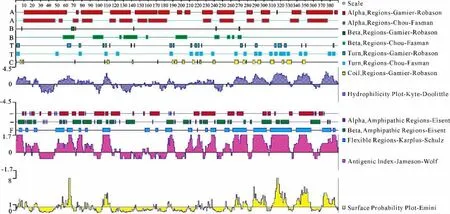
图2 PPRV病毒N蛋白的二级结构分析结果Fig.2 Analysis results of secondary structures of N protein in PPRV
2.5 PPRV间接ELISA检测方法的初步建立
2.5.1 人工合成多肽筛选结果 按方法1.2.5,分别以人工合成的4段多肽作为包被物,对血清样品进行间接ELISA检测。经筛选,确认编号为PPRVN(264-323)和PPRV-N(306-365)两段多肽与小反刍兽疫病毒标准阳性血清的反应特异性良好,与标准阴性血清、其他几种常见山羊病阳性血清无交叉反应。与进口标准竞争ELISA试剂盒同时检测系列稀释的PPRV标准阳性血清,检测极限均可达1∶200,表明敏感性相当。做好的包被板在4℃存放,2周内使用效果无变化,在-20℃存放,3个月内使用效果无变化,表明这两段多肽作为ELISA包被物稳定性良好。
2.5.2 多肽联合包被物及阳性血清最佳稀释度的筛选 本研究选定检测效果良好的编号为PPRV-N(264-323)和PPRV-N(306-365)两段多肽作为联合包被物,分别以pH 9.2的碳酸盐缓冲液稀释成2μg/100μL~0.25μg/100μL包被反应液,两种多肽以1∶1、1∶2、1∶3、1∶4多种比例分别混合作为联合包被物,血清从1∶25到1∶200倍比稀释,酶标二抗1∶5 000倍稀释,与两组多肽单独作为包被物的情况进行比对检测试验(部分数据列于表1,表中的OD值数据为多次试验的平均值)。结果显示,随着血清稀释倍数的增加和两种人工多肽抗原包被总浓度的降低,血清的OD值随之降低,当血清稀释度在1∶100,两种多肽按1∶1比例混合成联合包被物且总浓度为1μg/100μL浓度时,阳性血清OD值接近于1.0,阴性血清OD值<0.2,并且阳性和阴性血清OD值的比值(P/N值)为最高,此时多肽联合包被物及阳性血清的稀释度为最佳工作条件。
2.5.3 间接ELISA判定标准的确定 按2.5.2筛选的条件,对小反刍兽疫病毒标准阴性血清和其他几种山羊病的参考血清进行ELISA检测,结果表明这些血清样品的OD值在0.156~0.284之间(具体数据略),样品平均值和标准差分别为=0.185,SD=0.038,得到置信区间上限+3SD=0.299≈0.30,将0.30作为阴性血清OD值的上限,根据统计学原理,当血清样品OD值>+3SD时,可以在99.9%的水平上判定为阳性。因此得出间接ELISA判定标准为,当待测样品的OD值大于0.30时判为阳性,小于或等于0.30时判为阴性。
2.6 临床血清样本检测的符合率
用所建立的间接ELISA检测方法和进口小反刍兽疫标准竞争ELISA试剂盒分别对山羊血清样品进行检测。本文建立的间接ELISA方法检出48份阳性,77份阴性;进口标准竞争ELISA试剂盒检出50份阳性,80份阴性(表2)。因此,研制的间接ELISA检测方法的诊断敏感性为96.0%,诊断特异性为96.25%,与进口标准竞争ELISA试剂盒的检测符合率为96.15%。
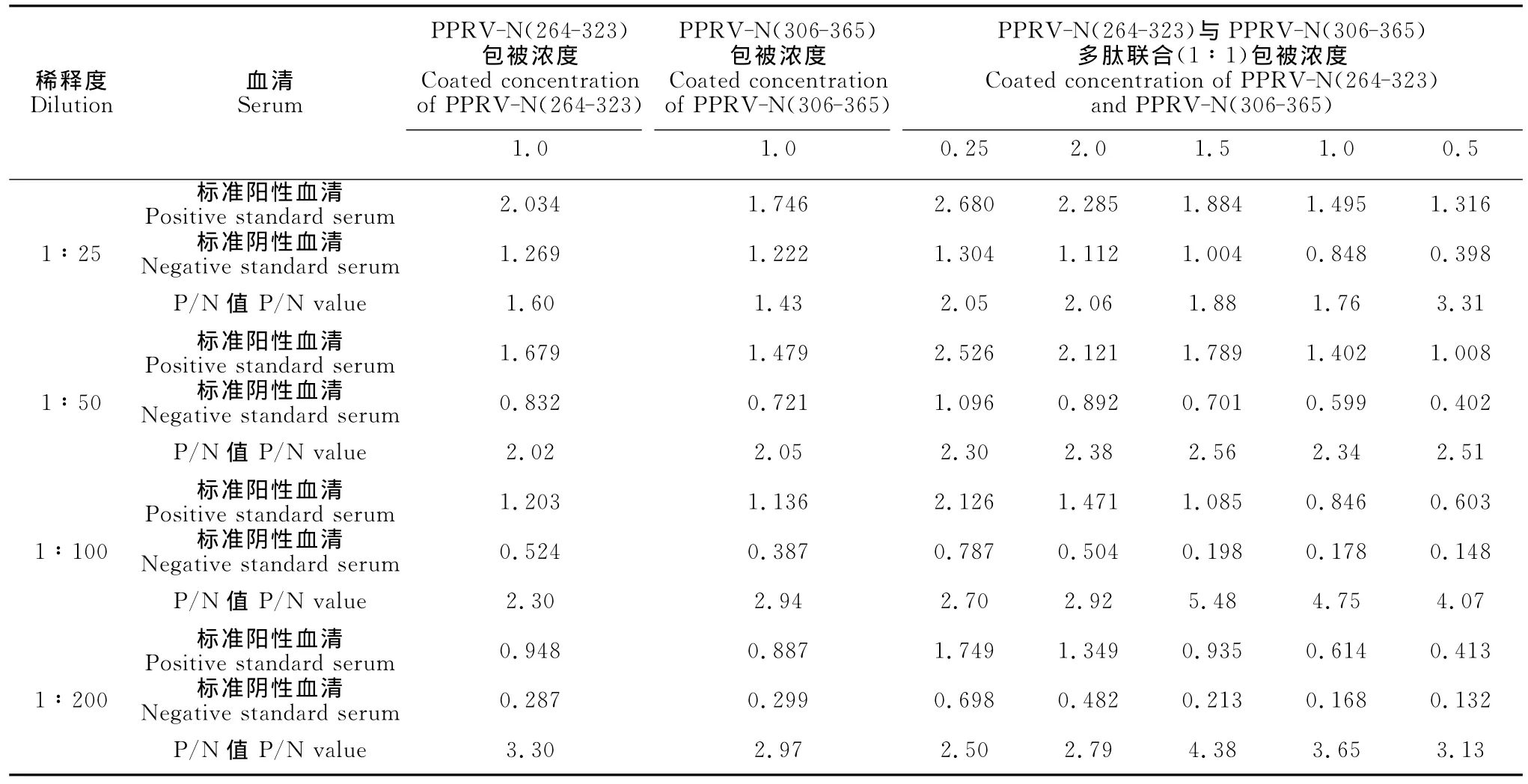
表1 两组人工多肽联合包被或单独包被ELISA比对检测试验结果Table 1 ELISA comparison test results of two artificial peptides coated together or separately μg/100μL

表2 自研的间接ELISA方法与进口标准竞争ELISA试剂盒比对试验结果Table 2 Comparison test results of iELISA homemade and the standard c-ELISA kit imported
(1)诊断特异性:77/80×100%=96.25%;
(2)诊断敏感性:48/50×100%=96.0%;
(3)符合率:(48+77)/130×100%=96.15%。
3 讨论
PPRV属于副黏病毒科的麻疹病毒属[1],与牛瘟病毒(Renderpest virus,RPV)最为接近,且PPR的临床症状和牛瘟相似,所以PPR又称为小反刍兽假性牛瘟[9]。已有报道指出,基于PPRV全长N蛋白的ELISA检测方法不能有效区分PPRV抗体和牛瘟病毒抗体[5,8],但靠近N蛋白C端的一段大小约为100个氨基酸的区域不保守,与麻疹病毒属其他成员之间的同源性仅为17%~30%[8],且为主要的免疫活性部位,可为特异性检测PPRV抗体提供最好的抗原[7-8,10,12]。
本文利用的多肽,是分子结构介于氨基酸和蛋白质之间的一类化合物,由一种或多种氨基酸按照一定的排列顺序通过肽键结合而成,既是构成蛋白质的结构片段,也是蛋白质发挥作用的活性基团[17]。通过对蛋白进行二级结构分析,寻找并确定B细胞抗原表位[11,13],设计尽可能短的多肽,不仅可以提高其免疫活性,减少交叉反应,还能实现多肽的高效合成,发挥其易于高纯度大量制备的最大特点[17]。多肽的这些优点克服了通过基因工程表达获得重组蛋白/多肽时需解决高效表达的技术难题、分离困难、产率低、成本昂贵和难以用于规模生产等局限性,多肽的应用已成为近年生命科学研究领域的一大热点[17]。
基于上述研究基础,本研究截取了PPRV N蛋白靠近 C端的2/3区段[8,12],进行了 B细胞抗原表位分析[11-12],获取了抗原结合位点的理论位置,设计了多肽序列并经化学合成得到多肽,用作包被物成功建立了PPRV血清抗体间接ELISA检测方法。该方法充分利用了人工合成多肽的生物特性和优势,应用到PPR的预防诊断技术的研发。人工多肽的设计,优势集中了B细胞抗原表位,提高了包被物与待检样品的结合几率,采用两段多肽作为联合包被物,血清抗体只要与其中一段多肽结合,就能显示阳性结果,可提高检测特异性和敏感性。与进口标准竞争ELISA试剂盒进行了比对,验证了研制的间接ELISA检测方法的有效性。
PPRV可通过直接接触进行水平传播,也可通过精液及胚胎等途径垂直传播,还可通过引种、国际贸易、野生动物迁徙等远距离跨界传播[1,6]。据OIE官网公布,至今我国已有22个省市出现了PPRV的感染病例,由于我国地域辽阔,南来北往人员流动大,动物及动物产品交易频繁,在缺乏有效的防控和预防诊断措施情况下,PPR在我国传播蔓延的速度仍然很快[3-4]。本文开创性地利用2段PPRV的N蛋白人工合成多肽,联合作为ELISA包被抗原,建立了PPRV血清抗体间接ELISA检测方法,与进口标准PPRV竞争ELISA试剂盒的检测符合性达96.15%,充实了我国PPRV诊断检测技术,也提供了动物疫病ELISA检测方法建立的新思路。
[1]蔡冬冬,李金海,邢 坤,等.小反刍兽疫概述[J].四川畜牧兽医,2014(5):31-33.
[2]郭君卫.小反刍兽疫疫情的防控[J].畜牧与饲料科学,2014,35(5):111-112.
[3]林春斌,涂凌云,吴录汉,等.我国小反刍兽疫现状和防控对策[J].江西畜牧兽医杂志,2014(3):47-48.
[4]朱迪国,宗建德,袁丽萍,等.全球小反刍兽疫流行趋势[J].中国动物检疫,2014,31(6):14-16.
[5]吴 宣,张永宁,关泽英,等.小反刍兽疫诊断技术的研究进展[J].四川畜牧兽医,2014(6):30-32.
[6]Padhi A,Ma L.Genetic and epidemiological insights into the emergence of peste des petits ruminants virus(PPRV)across A-sia and Africa[J].Sci Rep,2014(4):1-7.
[7]邱文英,李 伟,李 刚,等.小反刍兽疫N蛋白单克隆抗体的制备及竞争ELISA方法的建立[J].农业生物技术学报,2011,19(5):967-972.
[8]邱文英,李 刚,田康乐,等.小反刍兽疫病毒N蛋白主要抗原区域的原核表达及间接ELISA检测方法的建立[J].中国兽医科学,2012,2(5):483-487.
[9]王 净,李 刚,史利军.间接ELISA和竞争ELISA检测小反刍兽疫病毒抗体的比较[J].畜牧兽医学报,2014,45(2):333-336.
[10]Muniraju M,Munir M,Parthiban A R,et al.Molecular evolution of peste des petits ruminants virus[J].Emerg Infect Dis,2014,20(12):2023-2033.
[11]李 倩,姚淑霞.非洲猪瘟病毒VP73蛋白的B细胞表位预测[J].安徽农业科学,2008,36(18):7680-7682.
[12]Yu R,Fan X,Xu W,et al.Fine mapping and conservation analysis of linear B-cell epitopes of peste des petits ruminants virus nucleoprotein[J].Vet Microbiol,2015,175(1):132-138.
[13]肖 镜,付 瑞,贺争鸣,等.猴B病毒BVgD-多肽ELISA检测方法的建立[J].实验动物科学,2008,25(2):20-26.
[14]曾少灵,廖立珊,唐金明,等.非洲猪瘟病毒VP73蛋白在昆虫细胞中的表达与间接ELISA方法的建立[J].动物医学进展,2013,34(1):1-6.
[15]杨俊兴,曹琛福,曾少灵,等.牛病毒性腹泻病毒双单克隆抗体夹心ELISA检测方法的建立及初步应用[J].动物医学进展,2013,34(5):11-16.
[16]曹琛福,梁云浩,陶 虹,等.非洲猪瘟病毒p54基因的原核表达及其抗体的间接LEISA检测方法的建立[J].动物医学进展,2014,35(2):6-10.
[17]李永振,贺继东,彭政,等.多肽的合成与应用进展[J].化学与生物工程,2010,27(4):9-14.
猜你喜欢
温州医科大学学报(2019年4期)2019-04-28
中国乳品工业(2018年10期)2018-11-16
河南畜牧兽医(2017年20期)2018-01-19
中国免疫学杂志(2017年1期)2017-01-17
湖南畜牧兽医(2016年1期)2016-06-05
上海故事(2015年13期)2016-01-22
伴侣(2015年5期)2015-09-10
河南畜牧兽医(2015年13期)2015-08-15
畜牧兽医学报(2015年3期)2015-07-05
医学研究杂志(2015年6期)2015-07-01
