
N-羟甲基叔胺的乙酰解反应及其在DAPT合成TAT反应机理中的应用
2015-05-10 01:06:24万子娟
含能材料 2015年11期
邹 坡, 万子娟, 罗 军
(南京理工大学化工学院, 江苏 南京 210094)
1 引 言
1,3,5,7-四硝基-1,3,5,7-四氮杂环辛烷(HMX)是目前获得大规模应用、综合性能最好的单质炸药。1,3,5,7-四乙酰基-1,3,5,7-四氮杂环辛烷(TAT)是合成HMX的一个重要前体,由TAT制备HMX的方法以高收率、高纯度而备受关注[1-2]。TAT通常可由3,7-二乙酰基-1,3,5,7-四氮杂双环[3.3.1]壬烷(DAPT)在乙酸酐中酰解获得[3-4]。Lukasavage等[3]认为,DAPT酰解时桥亚甲基首先发生断裂,得到7-乙酰氧基亚甲基-1,3,5-三乙酰基-1,3,5,7-四氮杂环辛烷,随后发生水解得到活性中间体7-羟甲基-1,3,5-三乙酰基-1,3,5,7-四氮杂环辛烷,该活性中间体在反应体系中不稳定,容易脱去甲醛得到仲胺,仲胺继续酰化得到目标产物TAT。李清霞等[5]使用半制备高效液相色谱分离TAT合成过程中7-乙酰氧基亚甲基-1,3,5-三乙酰基-1,3,5,7-四氮杂环辛烷等稳定中间体,为TAT的形成机理提供了部分直接的证据。但是,由于反应体系复杂,活性中间体存在时间极短,无法通过常规的实验手段获得或捕获,这给证明反应机理带来了困难。本研究通过合成含N-羟甲基的叔胺来模拟DAPT乙酰解反应中可能存在的活性中间体7-羟甲基-1,3,5-三乙酰基-1,3,5,7-四氮杂环辛烷的乙酰解反应过程,试图找出一般规律,对TAT的合成进行解释; 并考察硝酸铵在N-羟甲基叔胺乙酰化中的作用,以期为3,7-二硝基-1,3,5,7-四氮杂双环[3.3.1]壬烷(DPT)硝解合成HMX中硝酸铵的作用机制提供一些依据。
2 实验部分
2.1 合成路线
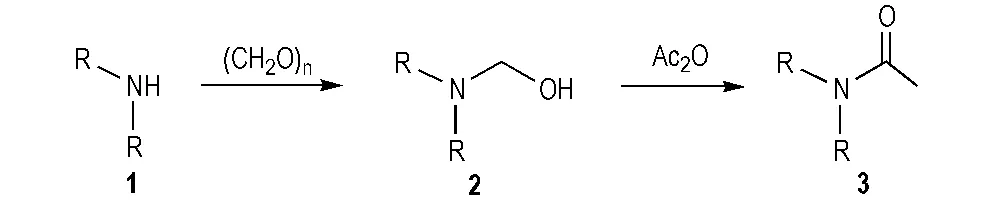
Scheme 1 Synthesis of compound 3
2.2 试剂与仪器
二异丙胺、二正丁胺、二烯丙胺(AR,阿拉丁试剂(上海)有限公司),吗啉(CP,阿拉丁试剂(上海)有限公司),乙酸酐(AR,上海凌峰化学试剂有限公司),甲醛溶液(AR,西陇化工股份有限公司),多聚甲醛、二氯甲烷、氯化钠、碳酸氢钠(AR,成都市科龙化工试剂厂),无水硫酸钠(AR,国药集团化学试剂有限公司)。
AVANCE III型500MHz核磁共振仪(瑞士Bruker公司),GC-9860气相色谱仪(上海奇阳信息科技有限公司),Trace 1300 GC-ISQ MS气相色谱-质谱联用仪(美国Thermo公司),Finnigan TSQ Quantum ultra AM 型质谱仪(美国Finnigan公司)。
2.3 仲胺的羟甲基化反应
链状仲胺的羟甲基化反应参照文献[6]方法进行:
将0.1 mol仲胺加入烧瓶中,缓慢滴入0.1 mol 37%的甲醛水溶液,充分搅拌。室温反应1 h后,升温至65 ℃继续反应2.5 h。反应结束后,向反应体系中加入一定量的氯化钠固体,搅拌溶解后冷却至室温,静置分层。分液除去下层水相,得到油状液体。通过核磁共振对混合物的比例进行定量分析。
N-羟甲基二正丁胺(2a):1H NMR (500 MHz, CDCl3)δ=4.18 (s, 2H), 2.66~2.62 (m, 2H), 2.47~2.43 (m, 2H), 1.49~1.28 (m, 8H), 0.93~0.90 (m, 6H);13C NMR (125 MHz, CDCl3)δ=83.5, 51.9, 29.4, 20.8, 14.1。
n(N-羟甲基二正丁胺)∶n(二正丁胺)=0.48∶0.52。
N-羟甲基二异丙胺(2b):1H NMR (500 MHz, CDCl3)δ=2.96~2.88 (m, 2H), 2.15 (s, 2H), 1.03 (d, 12H,J=6.6 Hz);13C NMR (125 MHz, CDCl3)δ=50.1, 31.3, 19.8; MS(ESI)m/z: 132 [M+1]+。纯物质。
N-羟甲基二烯丙胺(2c):1H NMR (500 MHz, CDCl3)δ=5.88~5.79 (m, 2H), 5.22~5.09 (m, 4H), 4.16 (s, 2H), 3.31~3.26 (m, 2H), 3.16~3.11 (m, 2H);13C NMR (125 MHz, CDCl3)δ=136.0, 117.2, 82.5, 53.8。
n(N-羟甲基二烯丙胺)∶n(二烯丙胺)=0.33∶0.67。
吗啉的羟甲基化反应参照文献[7-8]方法进行:
将3 g(0.1 mol)多聚甲醛加入烧瓶中,冰浴保护下缓慢滴入8.71 g(0.1 mol)吗啉,剧烈搅拌。滴完吗啉后,撤去冰浴。室温反应2 h后,加热至60 ℃反应10 h。冷却至室温,加入CH2Cl2,有白色固体析出。过滤,收集滤液,减压蒸馏除去溶剂后得到无色粘稠液体。通过核磁共振对混合物的比例进行定量分析。
N-羟甲基吗啉(2d):1H NMR (500 MHz, CDCl3)δ=4.14 (s, 2H), 3.75~3.69 (m, 4H), 2.72~2.67 (m, 4H), 2.04 (br, 1H);13C NMR (125 MHz, CDCl3)δ=87.3, 67.0, 49.8。
二吗啉甲烷(2d′):1H NMR (500 MHz, CDCl3)δ=3.75~3.69 (m, 8H), 2.91(s, 2H), 2.50(br, 8H);13C NMR (125 MHz, CDCl3)δ=81.6, 67.0, 52.0。
n(N-羟甲基吗啉)∶n(二吗啉甲烷)=0.47∶0.53。
2.4 N-羟甲基叔胺的乙酰解反应
将仲胺与N-羟甲基叔胺的混合物(共5 mmol)和乙酸酐(6 mmol)依次加入到烧瓶中,搅拌均匀,加热至85 ℃反应2 h[9]。反应结束后,待体系冷却至室温,加入CH2Cl2萃取。有机相用饱和NaHCO3溶液洗涤至中性,再经无水硫酸钠除水后,过滤,滤液减压蒸馏除去溶剂得到红棕色液体。所得产物经1H NMR、13C NMR检测,并与文献对比确定。
N,N-二正丁基乙酰胺(3a)[10]:1H NMR(500 MHz, CDCl3)δ=3.30 (t, 2H,J=7.7 Hz), 3.21 (t, 2H,J=7.7 Hz), 2.09 (s, 3H), 1.58~1.47 (m, 4H), 1.37~1.27 (m, 4H), 0.95 (t, 3H,J=7.4Hz), 0.92 (t, 3H,J=7.4Hz);13C NMR (125 MHz, CDCl3)δ=170.2, 48.6, 45.5, 31.1, 29.9, 21.5, 20.3, 20.1, 13.9, 13.8。
N,N-二异丙基乙酰胺(3b)[11]:1H NMR (500 MHz, CDCl3)δ=3.92~3.87 (m, 1H), 3.53 (br, 1H), 2.08 (s, 3H), 1.37 (d, 6H,J=6.8 Hz), 1.21 (d, 6H,J=6.8 Hz);13C NMR (125 MHz,CDCl3)δ=169.6, 49.3, 45.5, 24.0, 21.0, 20.6。
N,N-二烯丙基乙酰胺(3c)[12]:1H NMR (500 MHz, CDCl3)δ=5.80~5.71 (m, 2H), 5.22~5.10 (m, 4H), 3.98 (d, 2H,J=6.0 Hz), 3.87~3.85 (m, 2H), 2.10 (s, 3H);13C NMR (125 MHz,CDCl3)δ=170.7, 133.3, 132.7, 117.3, 116.6, 50.0, 47.8, 21.4。
N-乙酰基吗啉(3d)[13]:1H NMR (500 MHz, CDCl3)δ=3.68 (q, 4H,J=5.3 Hz), 3.62 (t, 2H,J=4.8 Hz), 3.47 (t, 2H,J=4.8 Hz), 2.10 (s, 3H);13C NMR (125 MHz, CDCl3)δ=169.2, 66.8, 66.6, 46.6, 41.7, 21.1。
3 结果与讨论
3.1 二正丁胺及其羟甲基衍生物的乙酰化反应
取5 mmol的底物(二正丁胺及其羟甲化衍生物)加入6 mmol的乙酸酐中于85 ℃反应2 h,考察硝酸铵对反应的影响,结果如表1所示。
表1 二正丁胺与N-羟甲基二正丁胺的乙酰化结果
Table 1 Acetylation of dibutylamine andN-hydroxymethyldibutylamine

entrysubstrateNH4NO3/mmolyield/%2)1-83258531)-10041)573
Note: 1) The molar ratio of 1a and 2a is 52∶48. 2) Isolated yields.
从表1中可以看出,对于二正丁胺的乙酰化,硝酸铵的加入与否对反应结果无明显影响。若有N-羟甲基二正丁胺存在,无硝酸铵时,乙酰化产率可达到100%,说明N-羟甲基二正丁胺进行乙酰化活性高于相应的二正丁胺,这与理论上叔胺氮原子碱性大于相应仲胺的分析一致。另外,用1H NMR和GC-MS对混合物的乙酰化反应进行跟踪,结果显示反应只生成一种物质N,N-二正丁基乙酰胺(N-酰化产物),并没有检测到羟基乙酸酯(O-酰化产物)。说明乙酰基是直接进攻氮原子,而没有进攻氧原子发生酯化反应。这一点也可以被仅用过量20%摩尔当量的乙酸酐就可以得到100%产率的结果所证明。但是,有N-羟甲基二正丁胺存在时,加入硝酸铵会使得乙酰化产物的收率明显降低,说明硝酸铵对N-羟甲基二正丁胺的乙酰解反应具有抑制作用。
3.2 底物的拓展
为研究不同底物对乙酰化反应的影响,取5 mmol的底物加入到6 mmol(反应4为7.5 mmol)的乙酸酐中于85 ℃反应2 h,结果如表2所示。
从表2中可以看出,不同取代基的底物在乙酰化中都脱去了羟甲基,得到相应的N-乙酰化产物。加入当量的硝酸铵后,酰化产物的收率出现不同程度的降低。说明该硝酸铵对不同结构N-羟甲基叔胺的乙酰解反应的影响规律具有普遍性。在所有实验中均发现,室温条件下硝酸铵在乙酸酐中的溶解性很差,没有N-羟甲基叔胺存在时,直到反应结束硝酸铵也无明显溶解; 而有N-羟甲基叔胺时,反应结束后硝酸铵全部溶解,而且体系中分离出了仲胺硝酸盐。这可能是因为硝酸铵与N-羟甲基叔胺酰解脱下的甲醛之间发生了反应,生成了可溶性物质,并放出硝酸与仲胺成盐,仲胺硝酸盐的酰化活性比自由胺小,不利于乙酰化反应的进行。
表2 不同N-羟甲基叔胺的乙酰解反应
Table 2 Acetolysis ofN-hydroxymethyldialkylamines

entrysubstrateproductyield1)/%yield2)/%11a+2a(52∶48)1007322b512431c+2c(67∶33)100594(47:53)8769
Note: 1) Isolated yields. 2) In the presence of NH4NO3(5 mmol), isolated yields.
3.3 温度对乙酰化结果的影响
为了更好地将实验结果应用到TAT的合成机理的解释上,用结构与活性中间体7-羟甲基-1,3,5-三乙酰基-1,3,5,7-四氮杂环辛烷更为相似的吗啉与甲醛的缩合产物为底物,将5 mmol的N-羟甲基吗啉和二吗啉甲烷的混合物加入到7.5 mmol的乙酸酐中反应2 h,考察了温度对酰化反应的影响,实验结果见表3。
表3 温度对乙酰化反应的影响
Table 3 Effect of reaction temperature on acetylation reaction

entrytemperature/℃reactionsystemyield/%3)10(CH3CO)2O58225(CH3CO)2O66325(CH3CO)2O—CH2Cl1)263425(CH3CO)2O—NH4NO2)364585(CH3CO)2O87
Note: 1) CH2Cl2(10 mL). 2) NH4NO3(5 mmol). 3) Isolated yields.
从表3中可以看出,温度对反应结果影响不大。脱羟甲基在低温也可顺利进行,随着反应温度的提高,酰化收率也有所增加。加入溶剂后,反应物的浓度降低,但收率几乎没变化,说明该反应的活性很高。室温时有无硝酸铵存在并不影响反应结果,可能是由于室温时硝酸铵不能与反应过程中产生的小分子反应,因此起不到抑制作用。
3.4 乙酰化反应机理
从结构来看,N-羟甲基叔胺有氮原子和氧原子两个亲核中心,因此与乙酸酐进行乙酰化反应存在两种可能性(Scheme 2): 以氮原子为进攻中心(进攻方式a)或以氧原子为进攻中心(进攻方式b)。如果以氮原子为进攻中心,则首先生成鎓盐中间体4,然后羟甲基正离子离去得到N-乙酰化产物3,羟甲基正离子分解得到甲醛; 如果以氧原子为进攻中心,则首先完成酯化反应离去乙酸分子生成化合物5,然后再以氮原子发生酰化反应得到N-乙酰化产物3和二乙酸亚甲酯。但是从理论上分析,叔胺氮原子的亲核性大于伯醇氧原子,因此按进攻方式a完成乙酰化的可能性更大。事实上,在二正丁胺和N-羟甲基二正丁胺的乙酰化反应,用1H NMR和GC-MS对反应进行跟踪,发现乙酰基是直接进攻氮原子,而没有进攻氧原子发生酯化反应。

Scheme 2 Mechanism of the acetolysis
为进一步证实上述反应过程,设计了两组实验: (1)以二烯丙基胺的羟甲基化产物为原料,在乙酸酐-硝酸铵中反应,每隔0.5 h取一次样,用薄层色谱法(TLC)跟踪,分析反应体系的组分; (2)参考文献[14],制备二乙酸亚甲酯,与反应体系中的组分进行对比。
TLC跟踪反应的结果显示,0.5 h时原料点消失,只能观察到N-乙酰化产物点。1,1.5 h和2 h的TLC结果与0.5 h时相同。加入硝酸铵后,N-乙酰胺收率会降低,但反应过程中未发现原料点。可以确定,硝酸铵并不参与氮原子的乙酰化反应。多聚甲醛在乙酸酐中合成二乙酸亚甲酯的后处理与N,N-二烷基乙酰胺一致,但在N-羟甲基酰化目标产物的核磁共振谱中并未发现二乙酸亚甲酯的存在。另外,将N,N-二正丁胺与N-羟甲基二正丁胺的混合物酰化后不处理直接取样检测,1H NMR数据显示混合物中除了N,N-二正丁基乙酰胺外还有乙酸,摩尔比为1∶1。
加入硝酸铵后仍按这种方法检测,除酰化目标产物、乙酸外,混合物中还存在一种离子盐。该离子盐含有二正丁胺的结构,经分析为二正丁铵硝酸盐。二正丁铵硝酸盐中的季氮原子不具有被乙酰化的活性,因此硝酸铵的加入可能具有两个作用: 一是与乙酰化反应产生的甲醛发生缩合反应生成乌洛托品及其类似物等可溶性化合物; 二是硝酸铵参与反应后放出的硝酸与仲胺成盐,从而抑制进一步乙酰化反应的进行。而这两个推论都被实验现象和结果支撑: 在仲胺及其羟甲基化产物N-羟甲基叔胺的混合物进行乙酰化反应的过程中,硝酸铵会随着反应进行逐渐消失,而且乙酰化产率降低,体系中有仲胺硝酸盐存在。
综上所述,实验结果证明氮原子的亲核性大于伯醇氧原子,乙酰化反应按进攻方式a完成。
3.5 DAPT乙酰解制备TAT的合成机理
上述研究结果可以用于解释DAPT乙酰解合成TAT的反应机理中。研究表明,DAPT在无水条件(乙酸酐体系)反应时,产率很低,通常使用10倍当量的乙酸酐、过高的温度以及催化剂来提高产率; Lukasavage等[3]将DAPT在乙酸酐-水-金属氧化物中反应,可得到81%的TAT,发现水的加入对反应有利; 王建龙等[4]对该法进行改进,用水代替金属氧化物作为催化剂,产率在80%左右。王建龙等[4]还发现,若不添加任何催化剂,在110 ℃反应时,仅能得到20%的TAT; 当加入一定量的水后,收率可大幅度提高到74%。综合以上文献报道可见,水的加入对反应结果有极大的影响,能显著加快反应并提高产率。综合本研究结论和文献结果,证实了DAPT乙酰解过程中活性中间体为7-羟甲基-1,3,5-三乙酰基-1,3,5,7-四氮杂环辛烷(8),反应历程如Scheme 3所示。

Scheme 3 Proposed process to prepare TAT from DAPT
DAPT在反应中首先与乙酸酐发生开环加成得到7-乙酰氧基亚甲基-1,3,5-三乙酰基-1,3,5,7-四氮杂环辛烷(7)[3,5],氧原子上乙酰基的存在使得7-位氮原子的电子云密度下降,因此进一步发生乙酰化反应比较困难,要使反应顺利进行,需要10倍当量的乙酰酐,并将反应温度维持在110 ℃的高温。当加入水后,化合物7发生水解得到羟甲基化产物8,与之相联的氮原子亲核性增强,更易发生酰化反应。实验也发现,加水后酰化反应在30 ℃左右就可以顺利进行。
4 结 论
(1) 构建了N-羟甲基叔胺,研究其乙酰解反应。研究结果表明,含N-羟甲基结构的叔胺在进行乙酰化时,反应发生在氮原子而不是氧原子,脱去羟甲基得到相应的N-乙酰化产物; 对比相应仲胺的乙酰化反应结果,发现N-羟甲基的引入有利于乙酰化反应的进行。加入过量20%摩尔当量的乙酸酐可以100%收率得到乙酰化产物。
(2) 考察了硝酸铵对反应的影响,硝酸铵在乙酰解过程中与酰解副产物甲醛发生了反应生成了可溶性物质,释放的硝酸与仲胺反应生成硝酸盐,从而抑制了乙酰化反应。加入硝酸铵后,乙酰化收率降低了18%~41%。
(3) DAPT乙酰解合成TAT的反应过程中,DAPT先与乙酸酐发生开环加成得到7-乙酰氧基亚甲基-1,3,5-三乙酰基-1,3,5,7-四氮杂环辛烷,随后水解得到活性中间体7-羟甲基-1,3,5-三乙酰基-1,3,5,7-四氮杂环辛烷,活性中间体在乙酸酐中继续反应可以顺利地得到TAT。本研究结果对DPT硝解合成HMX中硝酸铵的作用及硝解反应机理研究也有一定的启发作用。
参考文献:
[1] Siele V I. Process for producing 1,3,5,7-tetraalkanoyl-1,3,5,7-octahydrotetra zocines: US, 3979379[P], 1976.
[2] 何志勇, 罗军, 吕春绪, 等. 1,3,5,7-四乙酰基-1,3,5,7-四氮杂环辛烷的硝解反应机理[J]. 含能材料, 2012, 20(4): 427-431.
HE Zhi-yong, LUO Jun, Lü Chun-xu, et al. Nitrolysis mechanism of 1,3,5,7-tetraacetyl-1,3,5,7-tetraza cyclooctane[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2012, 20(4): 427-431.
[3] Lukasavage W J, Behrmann L A, Voreck W E. Process for making an HMX product: WO, 0073285[P], 2000.
[4] 王建龙, 陈丽珍, 曹端林, 等. TAT的制备方法: CN, 101768127A[P], 2010.
WANG Jian-long, CHEN Li-zhen, CAO Duan-lin, et al. Preparation of 1,3,5,7-tetraacetyl-1,3,5,7-tetra azacyclooctane: CN, 101768127A[P], 2010.
[5] 李清霞, 王鹏, 娄忠良, 等. 1,3,5,7-四乙酰基-1,3,5,7-四氮杂环辛烷合成反应中间体的制备分离[J]. 有机化学, 2012, 32: 165-168.
LI Qing-xia, WANG Peng, LOU Zhong-liang, et al. Separation of key intermediates in the synthesis of 1,3,5,7-tetranitro-1,3,5,7-tetrazocine by semi-preparative high performance liquid chromatography[J].ChinJOrgChem, 2012, 32: 165-168.
[6] 王永斌, 吴斌, 李冠军, 等. 胺基甲基黄原酸氰乙酯化合物和制备方法及其捕收剂: CN 102702055A[P], 2012.
WANG Yong-bin, WU Bin, LI Guan-jun, et al. Preparation of xanthic acid derivatives as collecting agents for flotation of sulfide ore and oxide ore: CN, 102702055A[P], 2012.
[7] Netscher T, Mazzini F, Jestin R. Tocopherols by hydride reduction of dialkyl amino derivatives[J].EurJOrgChem, 2007, 7: 1176-1183.
[8] Turlinton M, Pu L.Preparation of (S)-3,3′-bis-morpholinomethyl-5,5′,6,6′,7,7′,8,8′-octahydro-1,1′-bi-2- naphthol[J].OrgSynth, 2010, 87: 59-67.
[9] Ranu B C, Dey S S, Hajra A. Highly efficient acylation of alcohols, amines and thiols under solvent-free and catalyst-free conditions[J].GreenChem, 2003 (5): 44-46.
[10] Li Y M, Ma L N, Jia F,et al. Amide bond formation through iron-catalyzed oxidative amidation of tertiary amines with anhydrides[J].JOrgChem, 2013, 78: 5638-5646.
[11] Chen C T, Kuo J H, Pawar V D. Nucleophilic acyl substitutions of anhydrides with protic nucleophiles catalyzed by amphoteric, oxomolybdenum species[J].JOrgChem, 2005, 70: 1188-1197.
[12] Autenrieth B, Frey W, Buchmeiser M R. A dicationic ruthenium alkylidene complex for continuous biphasic metathesis using monolith-supported ionic liquids[J].Chem-EurJ, 2012, 18: 14069-14078.
[13] Pelagalli R, Feroci M, Chiarotto I, et al. Isopropenyl acetate, a remarkable, cheap and acylating agent of amines under solvent- and catalyst-free conditions: a systematic investigation[J].GreenChem, 2012, 14: 2251-2255.
[14] Manjula K, Pasha M A. Rapid and efficient method for the synthesis of acylals from aldehydes and their deprotection catalyzed byp-toluene sulphonic acid (p-TSA)[J].SynthCommun, 2007, 37: 1563-1569.
猜你喜欢
北京大学学报(自然科学版)(2022年4期)2022-08-18 06:58:18
应用化工(2022年2期)2022-04-27 01:29:08
华南师范大学学报(自然科学版)(2021年2期)2021-04-23 05:05:36
云南化工(2019年9期)2019-11-11 06:54:42
色谱(2019年5期)2019-05-29 09:15:14
分析化学(2019年3期)2019-03-30 10:59:24
石油炼制与化工(2018年11期)2018-11-13 08:01:38
山东工业技术(2018年11期)2018-06-27 10:16:04
含能材料(2016年10期)2016-05-09 03:10:40
云南化工(2015年4期)2015-01-11 05:10:20
