
H PLC测定清洁验证残留物非诺贝特的含量
2015-04-07 06:04:26洪丽萍黄加秀蔡亚兰宗艳艳谢春娟
中国合理用药探索 2015年8期
洪丽萍 黄加秀 蔡亚兰 宗艳艳 谢春娟
(扬子江药业集团有限公司,江苏 泰州 225321)
H PLC测定清洁验证残留物非诺贝特的含量
洪丽萍 黄加秀 蔡亚兰 宗艳艳 谢春娟
(扬子江药业集团有限公司,江苏 泰州 225321)
目的:建立清洁验证中残留物非诺贝特含量测定的高效液相色谱法。方法:色谱柱为 Agilent ZORBAX SB-C18(150 mm×4.6 mm,5 μm),流动相为水(磷酸调节pH至2.5)-乙腈(30∶70),检测波长:286 nm,流速:1.0 mL/min,柱温:25℃,进样量:20 μL。结果:非诺贝特在0.09~ 0.90 μg/mL范围内线性关系良好,r2=0.999 4;回收率:99.69%,RSD=0.16%(n=9)。结论:该法操作简便、结果准确,可以用于清洁验证残留物非诺贝特的定量分析。
高效液相色谱法;清洁验证;残留物;非诺贝特
非诺贝特是降低三酰甘油的首选药物之一[1],为第2代苯氧芳酸类药物,与吉非贝齐、苯扎贝特同属于贝特类调脂药物,于1998年在美国上市。非诺贝特可显著降低三酰甘油(TG)、适度降低总胆固醇(TC)和低密度脂蛋白胆固醇(LDL-C)水平,并能升高高密度脂蛋白胆固醇(HDL-C)水平,发挥良好的调脂作用,因而在临床上得到了广泛应用。其制剂非诺贝特缓释片是扬子江药业集团有限公司生产的主打畅销品种,用于调血脂,适用于经适当和正规饮食疗法不能控制的高胆固醇血症和/或高三酰甘油血症,因其显著疗效而受到广大患者的一致好评。本研究主要围绕非诺贝特缓释片清洁验证中的残留物非诺贝特开展研究,本研究参考《中华人民共和国药典》[2]和原国家食品药品监督管理局标准YBH04472014对主要活性成分非诺贝特的含量方法进行了研究,通过对非诺贝特缓释片生产用设备清洁验证的检验,确定该方法可以用于该产品清洁验证残留物的检测,保证清洁验证的准确性,并对每个验证参数设立可接受标准,结果表明高效液相色谱法(HPLC)简便,准确,可以用于清洁验证残留物非诺贝特的定量分析。
扬子江药业集团有限公司生产非诺贝特缓释片的固体制剂生产车间,是根据《药品生产质量管理规范(2010年修订)》及可生产品种特性、工艺流程及相应法律制度要求,对厂房、生产设施和设备进行了合理设计和布局,设计为多品种共线生产。依据《药品生产质量管理规范(2010年修订)》第四十六条(为降低污染和交叉污染的风险,厂房、生产设施和设备应当根据所生产药品的特性、工艺流程及相应洁净度级别要求合理设计、布局和使用,并符合要求)规定,对生产安全和有效性进行风险评估,以期对生产安全风险能正确认识并采取降低安全风险意见的控制措施,使生产质量及风险降低到可以接受的水平。进行风险评估所用的方法遵循失效模式与影响分析技术(FMEA),包括:①分析药物活性成分(APIs)及活性物质溶解度是否为该生产线所生产品种中最低的,即是否为最难清洁的成份;②理论残留限度是否为参照物组中最低限;③根据①、②分析,得出初步结论,评估是否需要开展清洁验证。
根据《药品生产验证指南》,本次采用生物学活性限度,即最低日治疗剂量(MTDD)的1/1 000作为确定最低残留量的依据。清洁的目的是保证在使用产品B时,不出现A产品的生理作用。B产品每天服用次数多,安全性下降,因此,上述MTDD的l/1 000系指B产品最多日使用制剂数中允许A产品残存的量,不超过因服用B产品而带入体内的A产品的最低日治疗剂量的l/1 000。MTDD数据来自药品标签和使用说明书上的有关数据计算:
MTDD= 每次给药片(粒)数 ×每片有效成分含量 ×每日最少给药次数。
根据MTDD计算单位面积残留物限度的过程如下:
①将相关设备生产的所有产品列表,在表中相应位置填写MTDD(mg),最小生产批量B(kg),单个制剂的质量Uw(g)和每日最多使用制剂数Dd;
②计算设备内表面积SA(cm2);
③取最小批量B为计算参数;
④取对应产品单位制剂质量Uw和日最大使用成品(制剂数)Dd为计算参数;
⑤计算该批产品理论成品数U:
U=1 000 B/Uw;
⑥计算设备表面残留物限度Ld:
Ld= 允许残留物总量/SA;

公司共线产品相关信息见表1。公司共线产品化学残留计算结果见表2。
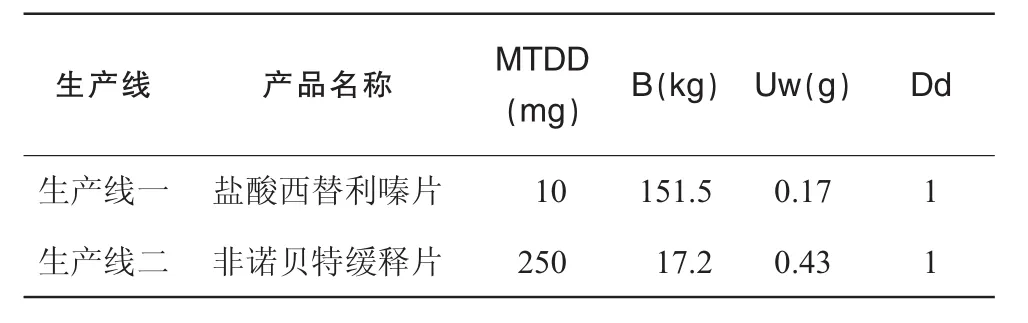
表1 共线产品信息
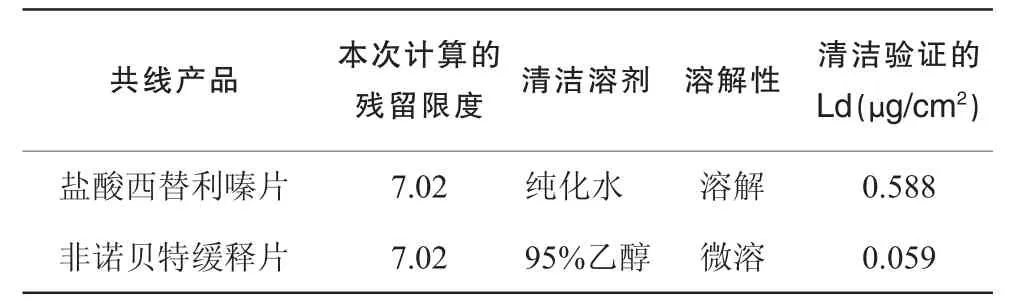
表2 公司共线产品化学残留计算结果
经对以上所列2个产品进行风险评估,盐酸西替利嗪片的设备与非诺贝特缓释片共用,清洁方法较非诺贝特缓释片易清洁,能满足限度要求,无交叉污染的风险,因而需对非诺贝特缓释片进行清洁验证,并对残留物非诺贝特进行检测以确定残留量是否超过限度,从而评价清洁是否有效,找到准确可靠的定量检测非诺贝特残留量的方法并通过对检测方法进行验证考察,可确认目前扬子江药业集团有限公司采取的防止污染和交叉污染、防止混淆与差错的措施适用于公司日常生产操作,可以确定该公司固体制剂生产车间用于多产品的生产是可行的,无交叉污染的风险。
DES都是通过氢键受体和氢键供体通过氢键连接成的混合盐溶液。最常用的氢键受体是季铵盐类的氯化胆碱,氯化胆碱价格低廉、可生物溶解、安全无毒,可以从生物质能源或者化石能源中提取。氯化胆碱可以与尿素、丙三醇、多元醇等安全廉价的氢键供体结合形成DES(见图5)。
非诺贝特缓释片生产用设备清洁验证的效果评价是通过HPLC进行的,采用擦拭法取样。
擦拭取样的残留量=设备中的总残留限度/设备总内表面积,
计算得出非诺贝特缓释片清洁验证中残留物非诺贝特的残留限度L=0.059 μg/cm2,试验中模拟设备材质的平板面积为100 cm2,溶于10 mL的乙醇中,其Ld值为0.59 μg/mL,因此以0.59 μg/mL作为残留物非诺贝特的取样回收率试验浓度。
1 试验仪器与试验样品
瑞士 Mettler Toledo XP205电子天平,美国GF315振荡器,美国安捷伦公司的Agilent1260高效液相色谱仪。
非诺贝特对照品(批号:100733-200401,含量:100.0%,来源:中国食品药品检定研究院);乙醇(AR,批号:20141023,厂家:国药集团化学试剂有限公司);磷酸(AR,批号:20140305,厂家:国药集团化学试剂有限公司);乙腈(HPLC,批号:1734530416,厂家:默克公司)。
2 方法与结果
2.1 色谱条件
流动相配制:水(磷酸调节 pH至 2.5)-乙腈(30∶70);色谱柱:Agilent ZORBAX SB-C18(150 mm× 4.6 mm,5 μm);进样体积:20 μL;流速:1.0 mL/min;检测波长:286 nm。
2.2 线性关系试验
Y=52.956 X+ 0.344 9,
相关系数r2=0.999 4;结果表明该方法在0.09~ 0.903 8 μg/mL浓度范围内,线性关系良好,考虑进样体积为20 μL,表明该方法进样量在1.8~ 18 ng范围内线性关系良好。

表3 线性溶液的配制

表4 线性溶液的配制
2.3 定量限
线性研究中合适浓度的溶液逐步稀释,确定定量限:移取线性检测项下L8溶液1 mL置于10 mL容量瓶中,配制成浓度为0.090 4 μg/mL的溶液,作为定量限检测溶液,结果定量限为 1.8 ng;S/N=10.3。
2.4 精密度试验
取浓度为 0.6 μg/mL的非诺贝特对照品溶液,按方法操作,重复测定6次,结果6次测定的RSD值为0.2%,表明该方法精密度良好。
2.5 稳定性试验
取浓度为 0.602 5 μg/mL的供试品溶液,在 25℃下放置考察,放置0,4,8,24 h之后分别测定1次,共测定4次,4次峰面积值的RSD值为0.5%,表明供试品溶液在24 h内稳定。
2.6 回收率试验
取已知含量的非诺贝特对照品溶液9份,分别精密加入一定量的非诺贝特对照品溶液(浓度为0.48,0.60,0.72 μg/mL)各3份,按上述色谱条件分别进样20 μL,根据测得峰面积值计算测得浓度,测得浓度与理论浓度之比即为回收率,计算RSD值,计算得平均回收率为99.69%,RSD为 0.16%,结果见表5。

表5 回收率试验结果(n=9)
3 结论
非诺贝特缓释片清洁验证中HPLC测定非诺贝特定量限为1.8 ng,即0.09 μg/mL。线性浓度范围为0.09~ 0.90 μg/mL;样品在室温下放置24 h内稳定。试验的验证结果表明HPLC测定清洁验证残留物非诺贝特的含量紫外光度法(UV)测定可行,该方法可用于非诺贝特的定量分析。
同时在清洁验证中需要注意:①要不断加强对员工在日常工作中的培训与管理,严格按照标准操作规程操作的意识和行为,确保有效的清洁和清洁状态维护。②在清洁验证工作中,需做好清洁验证中清洁效果的有效性及可行性确认工作,特别是对于新增的产品、改变批量的产品、变更设备等重要情况需要作好质量风险评估以确定验证内容的范围和深度以及在新的情况下生产车间多产品共用的可行性,重点放在检查防止污染和交叉污染、防止混淆与差错的措施并评估其适用性和有效性上。
[1] 李芳,俸灵林,阎超.非诺贝特制剂研究进展[J].世界临床医药,2011,32(9):552-557.
[2] 国家药典委员会.中华人民共和国药典(二部)[S].北京:中国医药科技出版社,2010.
Determination of Fenofibrate Residues in Cleaning Validation of Fenofibrate Sustained-release Tablets by HPLC
Hong Liping,Huang Jiaxiu,Cai Yalan,Zong Yanyan,Xie Chunjuan(Yangtz River Pharmacitical Group Co.,Ltd.,Jiangsu Taizhou 225321,China)
Objective:To establish a method for the content determination of fenofibrate residues in the cleaning validation of fenofibrate sustained-release tablets by HPLC.Methods:Separation was carried out using Agilent ZORBAX SB-C18(150 mm×4.6 mm,5 μm)column with phosphoric acid solution(pH=2.5)-acetonitrile(30∶70)as the mobile phase,the detection wavelength was at 286 nm,the flow was 1.0 mL/min and the column temperature was at 25℃ with sample size of 20 μL.Results:The linear range of fenofibrate concentration was from 0.09 to 0.90 μg/mL and the correlation coefficient was 0.999 4.The average recovery of samples was 99.69%and RSD was 0.16%(n=9).Conclusion:This method is convenient and accurate without interference and is suitable for the quantitative determination of fenofibrate residues in the cleaning validation of fenofibrate sustained-release tablets.
HPLC;Cleaning Validation;Residue;Fenofibrate
10.3969/j.issn.1672-5433.2015.08.007
2015-05-28)
洪丽萍,女,药师。研究方向:药物分析和标准研究。E-mail:hongliping@yangzijiang.com
猜你喜欢
西部交通科技(2022年2期)2022-04-27 00:34:29
食品安全导刊(2021年21期)2021-08-30 08:21:42
石油沥青(2019年6期)2020-01-16 08:57:16
山西化工(2019年1期)2019-03-28 11:57:58
大理大学学报(2018年2期)2018-04-21 07:00:38
校园英语·下旬(2018年12期)2018-02-26 12:48:32
山东医药(2017年5期)2017-04-05 05:18:49
东方考古(2016年0期)2016-07-31 17:45:44
华人时刊(2014年2期)2014-03-28 00:45:01
眼科新进展(2014年3期)2014-03-08 07:20:26
