
烯烃聚合用耐热型(α-二亚胺)镍催化剂的进展
2015-03-28 08:37:24臧丹丹范志强傅智盛
合成树脂及塑料 2015年2期
臧丹丹,何 峰,范志强,傅智盛
(高分子合成与功能构造教育部重点实验室,浙江大学高分子科学与工程学系,浙江省杭州市 310027)
在过去的30年中,研究人员致力于设计和开发均相的、单活性中心烯烃聚合催化剂。单活性中心催化剂的配体设计灵活多样,是研究催化机理、控制催化剂结构和裁剪烯烃聚合物结构的有效手段,是非均相催化剂体系无法实现的[1]。在这期间,最重大的进展之一是Brookhart等[2-3]开发了活性非常高的烯烃聚合用后过渡金属催化剂,用这些(α-二亚胺)镍/钯催化剂(见图1)可以制备高相对分子质量聚乙烯,可以耐受极性单体并实现乙烯与极性单体的共聚合[4-7],还可以通过调节乙烯压力制备线形到高度支化的聚乙烯,从而实现对聚乙烯拓扑结构的调控[8-10]。(α-二亚胺)镍/钯催化剂在温度不高于60.0 ℃时就会迅速分解,极大限制其工业化,因为工业生产中(尤其是气相聚合),通常的聚合温度为70.0~110.0 ℃[8,11];而且随着聚合温度的升高,所制聚乙烯的相对分子质量通常会下降。提高聚合温度一方面使聚合过程中的链转移反应(通过β-H消除反应)加速,另一方面降低乙烯在溶剂中的溶解度。因此,开发在较高聚合温度条件下稳定并可以制备高相对分子质量聚乙烯的(α-二亚胺)镍/钯催化剂,对其实现工业化意义重大。耐热型(α-二亚胺)钯催化剂的开发相对滞后,到目前为止,(α-二亚胺)钯催化剂催化乙烯均聚合或乙烯与极性单体的共聚合温度高于35.0℃的公开报道很少[12]。本文综述了耐热型(α-二亚胺)镍催化剂的研究进展,大致可以分成两类,一类是对与N相连的苯环进行修饰,另一类是对α-二亚胺的骨架进行修饰。
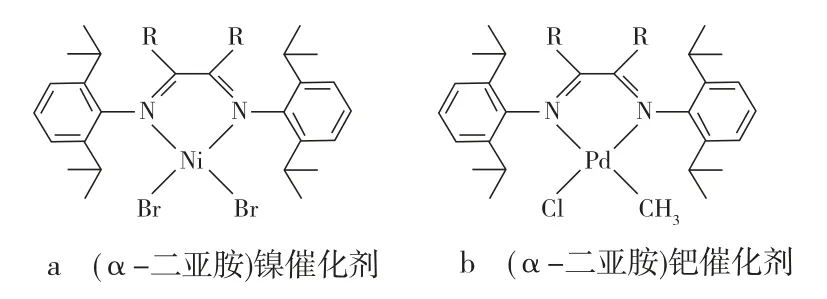
图1 典型(α-二亚胺)镍/钯催化剂结构示意Fig.1 Structure schematics of typical α-diimine nickel and palla dium catalysts
1 对与N相连的苯环进行修饰
Brookhart等[8]于1995年首次报道了能制备高相对分子质量聚乙烯的(α-二亚胺)镍/钯催化剂后,系统研究了聚合温度、乙烯压力以及配体结构对聚乙烯性能的影响,并提供了一种耐热型(α-二亚胺)镍催化剂。该催化剂甚至在85.0℃时仍表现出很高活性,聚合20 min时,活性为4.37×104kg/(g·h)。不过,聚乙烯数均分子量(Mn)仅为2.94×104,相对分子质量分布(Mw/Mn,Mw为重均分子量)为2.10,聚乙烯支化度很低,仅为19个/1 000 C,因此它的熔点高达121.0℃。另外,制备该催化剂的产率较低,为42%。
Guan Zhibin等[13]报道了一类与N相连的、基团为环芳基的(α-二亚胺)镍催化剂。该催化剂在90.0 ℃催化乙烯聚合时,表现出极高的活性(聚合15 min时,乙烯单体转化率为6.60×105mol/h),所制聚乙烯的相对分子质量高(Mn为29.20×104),Mw/Mn窄(为 1.40),支化度为96个/1 000 C;但随聚合时间的延长,该催化剂的活性衰减明显。此外,在制备该催化剂的配体时,需经三步合成,每步反应都需用过渡金属进行催化。因此,该催化剂的合成过程堪称合成的一个巧妙案例。
Ionkin等[14]开发的苯并呋喃基取代的η3-烯丙基(α-二亚胺)镍催化剂表现出极高的耐热性,在150.0 ℃时仍能以很高活性催化乙烯聚合。在60.0~70.0 ℃条件下可制备Mw超百万的聚乙烯,且Mw/Mn很宽(≥36.80)。用该催化剂制备的聚乙烯的支化度较低,熔点高于86.8 ℃。
Sun Wenhua等[15]报道了一系列含有大体积取代基的不对称(α-二亚胺)镍催化剂。该催化剂在80.0 ℃条件下能高活性地催化乙烯聚合,但所制聚乙烯的相对分子质量极低(Mw=1.62×104)。尽管其支化度高达166个/1 000 C,但熔点仍高达53.8 ℃。这可能和聚乙烯中几乎所有支链都是甲基有关,聚乙烯的链结构与乙烯-丙烯嵌段共聚物类似。另外,该催化剂只能用改性的甲基铝氧烷在n(Al)∶n(Ni)>1 000的条件下活化,廉价的一氯二乙基铝或倍半乙基氯化铝不能活化该催化剂。
Long等[16]用2,6-双(二苯基甲基)-4-甲基苯胺与二酮反应制备(α-二亚胺)镍催化剂。该催化剂催化乙烯聚合时,活性高,甚至在温度高达100.0 ℃时仍能制备相对分子质量较高(Mw=9.40×104)、Mw/Mn较窄(为 1.09~1.46)的聚乙烯。不过,聚乙烯的支化度不高(为63~75个/1 000 C),熔点高于37.0 ℃。
以上工作均通过修饰与N相连的苯环,在苯胺的2,6位引入大体积取代基,通过增加活性中心周围的空间位阻来达到提高催化剂热稳定性的目的;但大体积取代基的引入严重妨碍苯胺与二酮的缩合,造成这一步反应的产率非常低。另外,引入大体积取代基造成配体的体积过大,降低了金属中心的链行走能力,无法充分发挥(α-二亚胺)镍催化剂链行走的特性,制备的聚合物均有明显熔点,无法用作弹性体。
2 对α-二亚胺的骨架进行修饰
2009年,Wu Qing等[17]首先尝试对α-二亚胺的骨架进行修饰,将常用的萘基或二甲基改成了崁基,而与N相连的基团仍为常用的2,6-二异丙基苯基,由此制备了一种新型的(α-二亚胺)镍催化剂。该催化剂在80 ℃时表现出适度活性(乙烯单体的转化率为1.26×104mol/h),所制聚乙烯的相对分子质量较高(Mn= 12.30×104),Mw/Mn较窄(为2.00)。更重要的是,聚乙烯的支化度较高(129个/1 000 C),聚乙烯完全是非晶态的,不再有熔点,而是出现了一个较低的玻璃化转变温度(tg,-53.0 ℃)。这说明对催化剂的骨架进行修饰也可以达到提高催化剂热稳定性的目的,而且保留了(α-二亚胺)镍催化剂的链行走特性。不过,该催化剂在制备过程中需要用到三甲基铝,催化乙烯聚合时,需要用甲基铝氧烷活化,而且n(Al)∶n(Ni)较高。
典型的(α-二亚胺)镍催化剂在60.0~80.0 ℃催化乙烯聚合时表现出很好的链行走能力,所制聚乙烯的tg低于-60.0 ℃(接近乙丙橡胶的tg),只是该催化剂活性很低,聚乙烯的相对分子质量也很低。与Wu Qing等开发的催化剂相比,典型的(α-二亚胺)镍催化剂骨架上的萘基比崁基小,链行走能力强,导致所制聚乙烯的tg较低。
3 热稳定性机理初探
Brookhart等[18]采用低温核磁共振波谱仪研究了(α-二亚胺)钯催化剂的失活机理。他们认为(α-二亚胺)配体上的芳基会围绕N—C旋转,当芳基上的烷基旋转到与α-二亚胺五元环共平面时,烷基上的H会和钯发生相互作用,通过C—H活化形成稳定的六元环而失活。
Brookhart等还研究了(α-二亚胺)镍催化剂失活的机理,对比了图2所示的催化剂2和催化剂2'[13]。催化剂2中与N相连苯环的6位没有烷基,而催化剂2′中与N相连苯环的6位有甲基。按照(α-二亚胺)钯催化剂失活的机理来推断,催化剂2′的热稳定性应该比催化剂2差,因为后者的苯环上没有烷基,苯环在旋转过程中不会通过H与金属中心发生相互作用而失活;但实验结果却正好相反,催化剂2的热稳定性比催化剂2′差。因此,Brookhart等认为,尽管还不清楚(α-二亚胺)镍催化剂的失活机理,但它的失活机理和(α-二亚胺)钯催化剂的不同,即(α-二亚胺)镍催化剂失活与苯环的2,6位存在烷基与否无关。
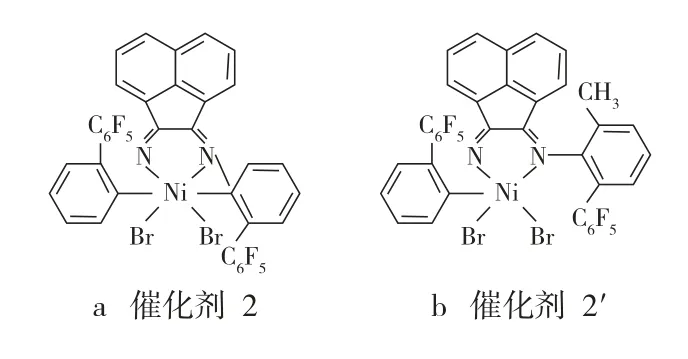
图2 催化剂2和催化剂2′的结构示意Fig.2 Structure schematics of the catalyst 2 and catalyst 2′
另外,Rieger等制备的三联芳基(α-二亚胺)镍/钯催化剂(见图3)在热稳定性方面并没有表现出过人的地方,尽管它们中与N相连苯环的2,6位均没有烷基。催化剂3在室温条件下活性不错,但Rieger等并没有发现它在较高温度时仍可以保持令人满意的活性;而催化剂3′在室温条件下催化乙烯聚合时几分钟就分解了。尽管催化剂4[12]中与其中一个N相连苯环的2,6位仍然保留了异丙基,但催化剂4表现出意想不到的热稳定性,它甚至在80.0 ℃还能催化乙烯聚合。
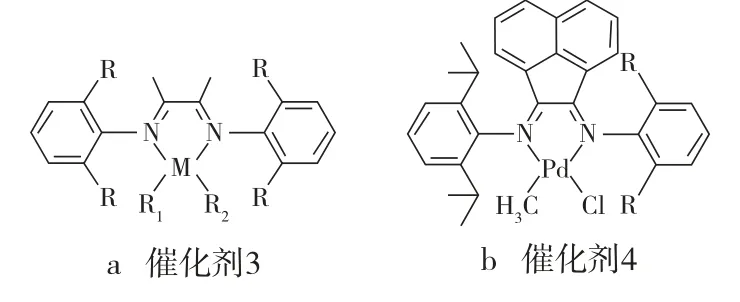
图3 催化剂3、催化剂3′和催化剂4的结构示意Fig.3 Structure schematics of the catalyst 3,catalyst 3′ and catalyst 4
经过单晶结构对比(见图4)发现,催化剂4的(α-二亚胺)五元环各边长(各元素间的键长)均相应增加,即催化剂4的五元环框架稍大[12]。这样使亚胺上取代基对钯的排斥力较小。进一步计算可知,催化剂4和催化剂3′中,钯到C1,C2的距离分别为0.277 8 nm和0.269 0 nm,即催化剂4中钯离配体的骨干较远,周围的空间较开阔,受配体的排斥力较小。这可能是催化剂4可以在溶液中稳定存在,并在较高温度时仍能催化乙烯聚合的主要原因。
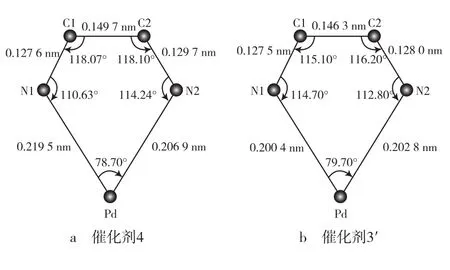
图4 (α-二亚胺)钯催化剂五元环的结构示意Fig.4 Structure schematics of the five-membered ring in the α-diimine palladium catalyst
因此,影响(α-二亚胺)镍/钯催化剂热稳定性的因素除了与和N相连苯环2,6位上的烷基有关,还与金属中心周围的空间结构有关。前人一味地通过向α-二亚胺配体的苯环或骨架上引入大体积取代基,并用大体积取代基阻碍苯环旋转,从而避免金属中心通过C─H活化而失活来解释所制催化剂的热稳定性,现在看来并不合理。
4 结语
(α-二亚胺)镍催化剂具有独特的链行走能力,因此,用其催化乙烯均聚合就可以制备含大量支链的聚乙烯,这样的聚乙烯具备用作弹性体的潜力。然而,要实现(α-二亚胺)镍催化剂在工业上的推广和应用,首先,要解决的是这类催化剂的热稳定性问题,其次,要弄明白影响这类催化剂热稳定性的决定性因素,避免催化剂设计过程中的盲目性。在保证催化剂具有良好热稳定性的同时,还要兼顾催化剂的链行走能力,充分发挥这类催化剂的链行走特性,使其在与现有的烯烃催化剂(如茂金属、MgCl2负载的Ziegler-Natta催化剂等)的竞争过程中独树一帜,制备出生产成本更低、附加值更高的聚烯烃新品种。
[1] Gibson V C,Spitzmesser S K. Advances in non-metallocene olefin polymerization catalysis[J]. Chemical Reviews,2003,103(1):283-316.
[2] Johnson L K,Killian C M,Brookhart M. New Pd(Ⅱ)- and Ni(Ⅱ)-based catalysts for polymerization of ethylene and alphaolefins[J]. J Am Chem Soc,1995,117(23):6414-6415.
[3] Killian C M,Tempel D J,Johnson L K,et al. Living polymerization of α-olefins using NiⅡ-α-diimine catalysts. Synthesis of new block polymers based on α-olefins[J]. J Am Chem Soc,1996,118(46):11664-11665.
[4] Boffa L S,Novak B M. Copolymerization of polar monomers with olefins using transition-metal complexes[J]. Chemical Reviews,2000,100(4):1479-1494.
[5] Johnson L K,Mecking S,Brookhart M. Copolymerization of ethylene and propylene with functionalized vinyl monomers by palladium(Ⅱ)catalysts[J]. J Am Chem Soc,1996,118(1):267-268.
[6] Mecking S,Johnson L K. Wang Lin,et al. Mechanistic studies of the palladium-catalyzed copolymerization of ethylene and α-olefins with methyl acrylate[J]. J Am Chem Soc,1998,120(5):888-899.
[7] Popeney C S,Guan Zhibin. A mechanistic investigation on copolymerization of ethylene with polar monomers using a cyclophane-based Pd(Ⅱ)α-diimine catalyst[J]. J Am Chem Soc,2009,131(34):12384-12393.
[8] Gates D P,Svejda S A,Onate E,et al. Synthesis of branched polyethylene using(α-diimine)nickel(Ⅱ)catalysts:influence of temperature,ethylene pressure,and ligand structure on polymer properties[J]. Macromolecules,2000,33(7):2320-2334.
[9] Guan Zhibin,Cotts P M,McCord E F,et al. Chain walking:a new strategy to control polymer topology[J]. Science,1999,283(5410):2059-2062.
[10] Younkin T R,Connor E F,Henderson J I,et al. Scope of olefin polymerization nickel catalysts-response[J]. Science,2000,288(5472):1750-1751.
[11] Xie Tuyu,Mcauley K B,Hsu J C C,et al. Gas phase ethylene polymerization:production processes,polymer properties,and reactor modeling[J].Ind Eng Chem Res,1994,33(3):449-479.
[12] Pan Huijie,Zhu Liang,Li Jiewei,et al. A thermal stable α-diimine palladium catalyst for copolymerization of ethylene with functionalized olefins[J]. Journal of Molecular Catalysis A:Chemical,2014,390:76-82.
[13] Camacho D H,Salo E V,Ziller J W,et al. Cyclophane-based highly active late-transition-metal catalysts for ethylene polymerization[J]. Angew Chem Int Ed,2004,43(14):1821-1825.
[14] Ionkin A S,Marshall W J. Ortho-5-methylfuran- and benzofuran-substituted η3-allyl(α-diimine)nickel(Ⅱ)complexes:syntheses,structural characterization,and the first polymerization results[J]. Organometallics,2004,23(13):3276-3283.
[15] Liu Hao,Zhao Weizhen,Yu Jiangang,et al. Synthesis,charac terization and ethylene polymerization behavior of nickel dihalide complexes bearing bulky unsymmetrical alpha-diimine ligands[J]. Catal Sci Technol,2012,2(2):415-422.
[16] Rhinehart J L,Brown L A,Long B K. A robust Ni(Ⅱ)α-diimine catalyst for high temperature ethylene polymerization[J].J Am Chem Soc,2013,135(44):16316-16319.
[17] Liu Fengshou,Hu Haibin,Xu Ying,et al. Thermostable α-diimine nickel(Ⅱ)catalyst for ethylene polymerization:effects of the substituted backbone structure on catalytic properties and branching structure of polyethylene[J]. Macromolecules,2009,42(20): 7789-7796.
[18] Tempel D J,Johnson L K,Huff R L,et al. Mechanistic studies of Pd(Ⅱ)-α-diimine-catalyzed olefin polymerizations[J].J Am Chem Soc,2000,122(28):6686-6700.
猜你喜欢
中学化学(2022年5期)2022-06-17 16:51:48
茶叶(2020年4期)2020-12-31 08:44:14
高中数理化(2020年1期)2020-02-29 02:21:18
四川师范大学学报(自然科学版)(2018年2期)2018-04-28 02:21:08
作文评点报·作文素材小学版(2016年45期)2017-03-06 17:31:23
河南科技(2015年2期)2015-02-27 14:20:35
昌吉学院学报(2013年1期)2013-12-08 07:36:22
食品科学(2013年19期)2013-03-11 18:27:17
烹调知识(2012年4期)2012-11-27 17:06:07
长江蔬菜(2012年2期)2012-08-09 08:43:30
