
抗HMGB1单抗对小鼠脑缺血—再灌注损伤后IL-1βIL-6表达的影响
2015-03-25 05:30陶庆霞
济宁医学院学报 2015年5期
孙 恺 谢 楠 陶庆霞 王 翀
(1潍坊医学院2013级研究生,山东 潍坊261053;2徐州市中心医院,江苏 徐州221000;3济宁医学院附属济宁市第一人民医院,山东 济宁272011)
缺血性脑卒中(ischemic stroke,IS)已经成为世界范围内主要的公共健康问题,也是引发较高致残率及致死率的首要原因[1]。脑缺血后通过快速恢复缺血组织血液灌注的方式在可以挽救濒临死亡的脑细胞的同时,也会产生脑缺血-再灌注损伤。大量研究表明,炎症反应在脑缺血-再灌注损伤发病机制中扮演极为重要的角色[2]。作为炎症反应的关键调控因子,高迁移率族蛋白B1(high mobility group protein box 1,HMGB1)通过与其受体结合参与炎症反应[3]。因此,通过以HMGB1为靶点治疗脑缺血-再灌注损伤已经成为人们研究的热点。本文通过建立小鼠局灶性缺血-再灌注模型,观察使用抗 HMGB1单抗(anti-HMGB1 monoclonal antibody,mAb)前后小鼠神经行为学、病理形态及脑水肿程度的改变,并对比脑组织内IL-1β和IL-6mRNA表达的差异,进而探讨使用mAb治疗脑缺血-再灌注损伤的可能机制。
1 材料与方法
1.1 主要试剂及仪器
栓线购自北京西浓科技有限公司;HMGB1单克隆抗体购自Abcam公司;抗HMGB1单克隆抗体由美国范斯坦医学研究所提供;小鼠单抗对照IgG抗体购自北京百奥莱博科技有限公司;HMGB1ELISA试剂盒购自美国R&D System Inc;Trizol试剂盒和 RT-PCR kit(二步法)购自加拿大invitrogen公司,使用Primers 3软件设计IL-1β和IL-6PCR引物。
1.2 实验动物及分组
8~12周C57BL/6J小鼠48只,体重16~20g,由北京军区总医院附属八一脑科医院实验室提供。随机分为假手术组(sham组),脑缺血-再灌注组(I/R组),抗 HMGB1单克隆抗体组(mAb组)和单抗对照组(control IgG组)4组,每组12只,分别用于脑水肿评估、脑部HE染色、脑组织IL-Iβ和IL-6mRNA 表达的检测,sham 组和I/R组测定血清HMGB1的水平。
1.3 脑缺血-再灌注模型制备
大脑中动脉闭塞(middle cerebral artery occlusion,MCAO)模型参考Zea Longa法经适当改良后制备。小鼠称重后腹腔注射3.6%水合氯醛(350mg/kg),显微镜下分离右侧颈总动脉(CCA),颈外动脉(ECA)和颈内动脉(ICA),充分游离ECA并于其在残端剪一小口,将栓线向内上方插入ICA颅内段,当遇到轻微阻力时停止,结扎ECA根部丝线。确认无出血后,消毒并缝合皮肤,2h后取出栓线。假手术组不插入线栓,余步骤同I/R组。
1.4 血清HMGB1水平测定
再灌注24h后采用心脏穿刺获取小鼠血样,按照ELISA试剂盒说明检测各组血清样本中HMGB1含量。每个样本重复3个复孔,将HMGB1单抗包被于酶标板上,加入生物素化的抗HMGB1抗体和辣根过氧化物酶标记的亲和素,3者形成免疫复合物,洗去游离成分,加显色底物及终止液,以酶标仪450nm测定吸光度,通过绘制标准曲线求出样品中HMGB1浓度。
1.5 脑组织含水量测定
再灌注后24h处死小鼠,沿脑桥上界水平面切断并分离左右半球,放于分析天平上称取脑半球组织湿重后,立即放入100℃烤箱烘烤48h得到干重。脑半球含水量的百分比通过公式计算:脑半球含水量百分比=(湿重-干重)/湿重×100% 。
1.6 HE染色观察各组小鼠脑组织病理变化
4组各取3只小鼠,4%多聚甲醛心脏灌注后断头取脑,4μm连续切片,常规HE染色,光学显微镜下观察脑组织切片上HE染色所示的细胞形态学变化。
1.7 实 时 荧 光 定 量 PCR 测 定IL-1β 和IL-6mRNA的表达
采用Trizol两步法提取总RNA,采用Primer3引物设计软件设计引物,引物分别为:IL-1β(L5'-GTGGAACTTGAGGCCACATT-3';R5'-T G T GA CA AA AA TG CC TG GA A),IL-6 (L5'-C CG GA GA GG AG AC TT CA CAG-3';R5'-T CC AC GA TT TC CC AG AG AA C-3'),HPRT (L5'-C AA GC TT GC TG GT GA AA AG GA-3';R5'-T GA AG TA CT CA TT AT AG TC AA GG GC AT ATC-3')。PCR扩增条件:95℃预变性10min,95℃15s,60℃60s,72℃延伸1min,重复共40个循环,得到IL-1β、IL-6和内参 HPRT的Ct值,以HPRT内参照作为内标,算出IL-1β、IL-6的相对表达量,以2-△△Ct为指标。
1.8 统计学方法
采用SPSS17.0软件进行统计分析。
2 结果
2.1 血清HMGB1的水平
I/R组、sham组小鼠 HMGB1浓度分别为(5.353±1.489)ng/ml、(47.75±3.667)ng/ml,I/R组明显高于sham组,差异有统计学意义(t值为10.71,P<0.001)。见图1。
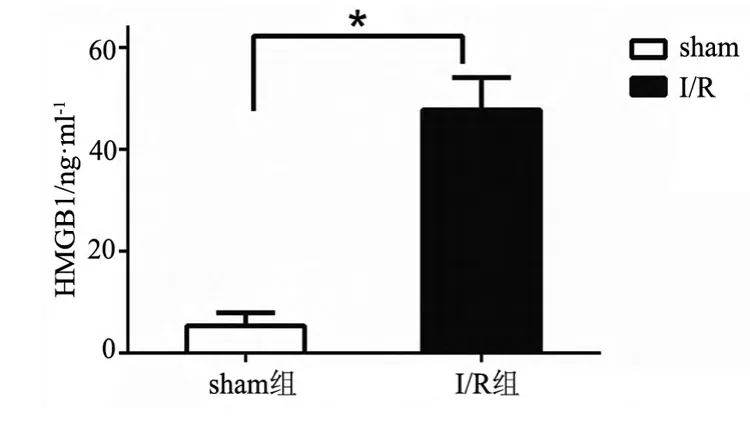
图1 脑缺血-再灌注组和假手术组小鼠血清HMGB1的水平
2.2 各组小鼠脑组织含水量的测定
干湿重法测得各组小鼠脑组织含水量的比较。I/R组、sham组、mAb组和control IgG组脑组织含 水 量 分 别 为 (78.34±2.836)%、(65.09±1.934)%、(67.47±1.775)% 和 (79.01±2.804)%,I/R组显著高于sham组,差异有统计学意义(t值为3.86,P<0.05),control IgG组显著高于mAb组,差异有统计学意义(t值为3.477,P<0.05),I/R组显著高于 mAb组,差异有统计学意义(t值为3.249,P<0.05)。见图2。
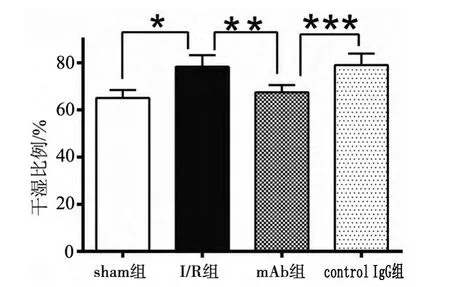
图2 各组小鼠脑组织含水量变化(%)
2.3 各组小鼠脑组织HE染色比较
sham组脑组织HE染色可见细胞及血管形态正常,结构完整,细胞间质均匀。I/R组脑组织神经元细胞数量明显减少,细胞形态结构不清,间质水肿明显,部分细胞出现坏死。IgG组与I/R组相比无明显变化,而mAb组脑组织较I/R组明显减轻,坏死细胞数量减少,可见部分细胞完整,细胞间质水肿程度减轻。
2.4 各组小鼠脑组织IL-1β和IL-6mRNA 的表达
利用实时荧光定量PCR技术测得各组小鼠IL-1β 和 IL-6mRNA 的 表 达 变 化。I/R 组 IL-1βmRNA的表达量 (0.783±0.028)明显高于sham组 (0.126±0.009),差异有统计学意义(t值为22.08,P<0.0001);mAb组 (0.417±0.047)与IgG组 (0.834±0.059)相比,前者表达量明显高于后者,差异有统计学意义(t值为5.5,P<0.01);I/R 组 IL-1βmRNA 表 达 量 (0.783±0.028)显著高于 mAb组 (0.417±0.047),差异同样有统计学意义(t值为6.681,P<0.01)。在IL-6mRNA 表达方面,I/R 组 (0.650±0.052)显著高于sham组 (0.059±0.009),差异有统计学意义(t值为11.6,P<0.001);而IgG组 (0.655±0.031)和 mAb组 (0.655±0.052)相比,mAb组(0.655±0.052)与I/R组 (0.650±0.052)相比,两组对比均无明显差异(t值分别为0.004和0.089,p值均>0.05)。见图3。
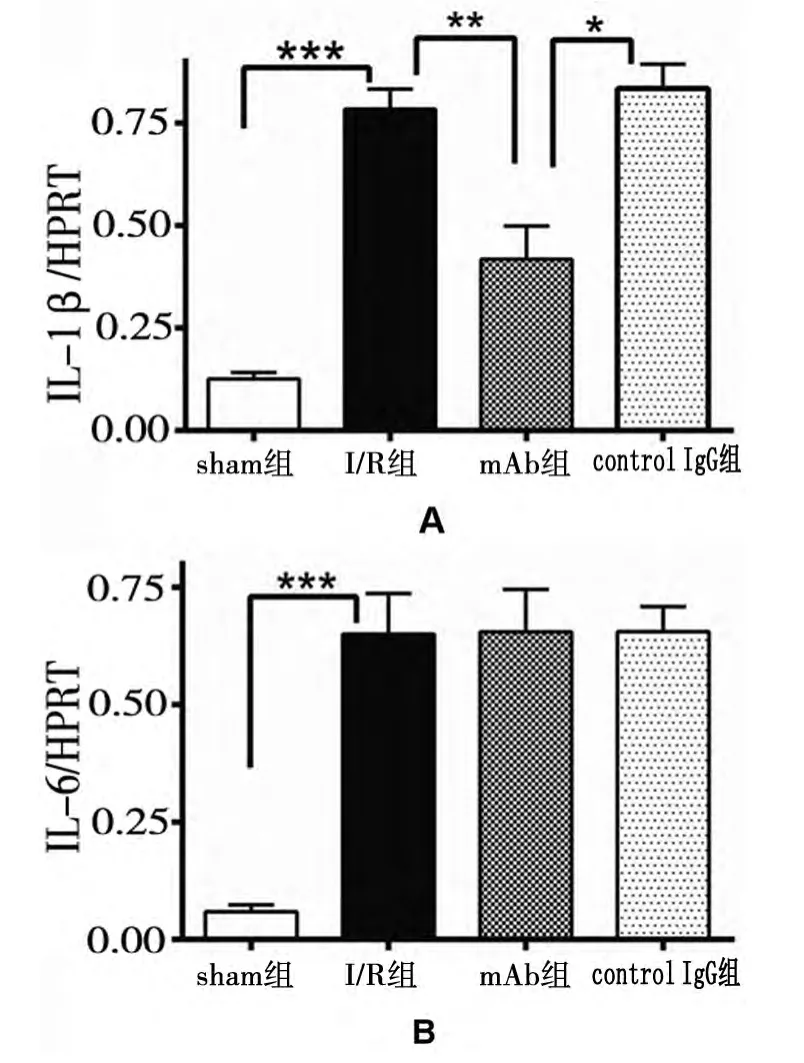
图3 各组小鼠IL-1β、IL-6mRNA 的表达变化
3 讨论
脑缺血-再灌注损伤通过一系列复杂的病理生理机制导致神经元细胞死亡以及神经功能障碍等脑损伤表现[4]。其中,炎症反应是参与上述复杂机制中较为重要的因素。脑缺血-再灌注时,在缺血灶局部存在大量炎症因子,并且炎症细胞的激活、浸润及黏附分子的合成分泌呈一种相互增强相互促进的级联反应,并通过一定的炎症信号通路使脑组织由缺血性损伤转向炎症性损伤[5]。
最初被发现是作为核蛋白与染色体结合参与维持核小体结构稳定以及调节基因转录等功能[6]。人们随后发现坏死的细胞可被动释放HMGB1至胞外[7];同时,激活后的巨噬细胞和胶质细胞等可主动分泌HMGB1[8]。释放至胞外的HMGB1具有促进炎症因子样的活性,通过与细胞膜表面受体如晚期糖基化终末产物受体(receptor for advanced glycation end products,RAGE)和 Toll样受体(toll like receptors,TLRs)结合,介导级联式的炎症反应[7]。在脑缺血-再灌注损伤病理机制中,HMGB1同样发挥着重要的作用。研究发现,小鼠MCAO模型及缺血性脑卒中患者脑组织中发现HMGB1由细胞核转移至细胞质,随后大量存在于脑脊液中[9]。本文结果显示,通过与假手术组对比,脑缺血-再灌注组小鼠血清内HMGB1的含量明显高于前者。胞外聚集的较高浓度的HMGB1通过募集单核/巨噬细胞并进一步激活星形细胞和小胶质细胞,从而促进炎症因子如IL-1β,IL-6,TNF-α等释放。本文通过对比脑缺血-再灌注组与假手术组小鼠脑组织IL-1β和IL-6mRNA的表达量,发现前者明显高于后者。说明HMGB1在参与脑缺血-再灌注损伤机制的同时,通过一系列复杂的级联式反应促进炎症因子的释放,从而进一步加重脑损伤。
目前,以HMGB1为治疗靶点的研究已经成为热点。通过采用与RAGE结合的HMGB1竞争性拮抗剂(如重组的HMGB1Box A功能域和由S100P衍生的RAGE),使用天然/合成的小分子HMGB1特异性抑制剂(如甘草酸和甲磺酸加贝酯),以及利用生物来源物质如HMGB1的特异性抗体、多肽、蛋白质或弯曲的DNA based双链等方式在抑制由HMGB1介导的相关疾病中发挥了重要作用[10]。此外,Liu等[11]发现,注射抗 HMGB1中和mAb可显著减轻脑梗死程度,表现为脑梗死体积减小90%,神经功能缺损评分明显提高,并减轻了血脑屏障的破坏。本文通过尾静脉注射mAb发现,脑水肿的程度显著减轻,由缺血-再灌注损伤引起的病理性破坏也得到一定程度的改善。作为神经毒性介质,IL-1β在脑缺血30min后即开始表达,并直接引发神经元细胞的凋亡并促进小胶质细胞和星形细胞过度表达炎症化学因子[12]。本实验结果显示,注射mAb后,IL-1βmRNA的表达量明显下降,提示抗HMGB1单抗的保护性作用。
然而,在本实验中,使用抗HMGB1单抗后,小鼠脑组织内IL-6mRNA的表达量并没有发生明显改变。我们推测,导致该结果出现的原因可能是由于IL-6在脑缺血-再灌注损伤过程中发挥双重作用有关。Huang等[13]研究发现,脑缺血-再灌注24h内,IL-6水平明显升高,可使基质金属蛋白酶-1(MMP-1)表达增高,促进细胞间黏附分子(ICAM)-1的表达,而导致脑组织损伤。然而,有研究显示,由脑细胞产生的IL-6通过激活STAT3信号通路发挥促血管生成及保护神经元细胞等功能[14-15]。本实 验 中,注 射 mAb后,IL-6 表 达 量 未发生明显变化,推测可能是其产生与mAb协同的保护性作用。
本文通过静脉注射抗HMGB1单克隆抗体观察及测定小鼠脑缺血-再灌注损伤后脑水肿程度,脑组织破坏程度以及脑组织内IL-1和IL-6mRNA的表达量的变化,表明HMGB1介导脑缺血-再灌注损伤机制中炎症反应过程,注射抗HMGB1单抗可有效保护受损脑组织。
[1] Bacigaluppi M,Comi G,Hermann D M.Animal models of ischemic stroke.Part one:modeling risk factors[J].Open Neurol J,2010,4:26-33.
[2] Shichita T,Sakaguchi R,Suzuki M ,et al.Post-ischemic inflammation in the brain[J].Front Immunol,2012,3:132.
[3] van Beijnum J R,Buurman W A,Griffioen A W.Convergence and amplification of toll-like receptor(TLR)and receptor for advanced glycation end products(RAGE)signaling pathways via high mobility group B1 (HMGB1)[J].Angiogenesis,2008,11(1):91-99.
[4] Kim J B,Sig Choi J,Yu Y M.HMGB1,a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain[J].J Neurosci,2006,26(24):6413-6421.
[5] 王萍,张密霞,庄朋伟,等.脑缺血再灌注损伤的炎症反应机制研究进展[J].天津中医药大学学报,2014,33(5):317-320.
[6] Bianchi M E,Agresti A.HMG proteins:dynamic players in gene regulation and differentiation[J].Curr Opin Genet Dev,2005,15(5):496-506.
[7] Scaffidi P,Misteli T,Bianchi M E.Release of chromatin protein HMGB1by necrotic cells triggers inflammation[J].Nature,2002,418(6894):191-195.
[8] Wang H,Bloom O,Zhang M,et al.HMG-1as a late mediator of endotoxin lethality in mice[J].Science,1999,285(5425):248-251.
[9] Kim S W,Lim C M,Kim J B,et al.Extracellular HMGB1Released by NMDA Treatment Confers Neuronal Apoptosis via RAGE-p38MAPK/ERK Signaling Pathway[J].Neurotox Res,2011,20(2):159-169.
[10]贺欣,贺丹,储小飞,等.通过调节高迁移率族蛋白B1治疗相关疾病的药物研究进展[J].药学服务与研究,2014,14(5):321-327.
[11]Keyue Liu,Shuji Mori,Hideo K.Anti-high mobility group box 1monoclonal antibody ameliorates brain infarction in-duced by transient ischemia in rats[J].FASEB J,2007,21(14):3904-3916.
[12]Takashi Shichita,Ryota Sakaguchi,Mayu Suzuki,et al.Postischemic inflammation in the brain[J].Front Immunol,2012,3:132.
[13]Huang J,Upadhyay U M,Tamargo R J.Inflammation in stroke and focal cerebral ischemia[J].Surg Neurol,2006,66(3):232-245.
[14]Jung J E,Kim G S,Chan P H.Neuroprotection by interleukin-6is mediated by signal transducer and activator of transcription 3and antioxidative signaling in ischemic stroke[J].Stroke,2011,42(12):3574-3579.
[15]Gertz K,Kronenberg G,Kälin R E,et al.Essential role of interleukin-6in post-stroke angiogenesis[J].Brain,2012,135(Pt 6):1964-1980.
猜你喜欢
实用肿瘤学杂志(2022年3期)2022-11-30
皮肤性病诊疗学杂志(2020年4期)2020-09-02
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
中成药(2017年6期)2017-06-13
中国卫生标准管理(2015年16期)2016-01-20
中国康复理论与实践(2015年10期)2015-12-24
吉林大学学报(医学版)(2015年4期)2015-12-17
中国体外循环杂志(2015年3期)2015-12-08
癌变·畸变·突变(2015年3期)2015-02-27
河南医学研究(2014年4期)2014-02-27
