
积雪草酸衍生物的合成与表征及其抗肿瘤活性研究
2015-03-22 05:18孟艳秋
沈阳化工大学学报 2015年3期
王 楠, 张 萌, 孟艳秋
(沈阳化工大学 制药与生物工程学院, 辽宁 沈阳 110142)
积雪草酸衍生物的合成与表征及其抗肿瘤活性研究
王 楠, 张 萌, 孟艳秋
(沈阳化工大学 制药与生物工程学院, 辽宁 沈阳 110142)
以积雪草酸为原料,对其C-2,C-3,C-23和C-28位基团进行结构修饰,设计并合成3类共10个化合物,并经IR、MS、1H NMR确证其结构.采用MTT法研究部分积雪草酸衍生物对人胃腺癌细胞株SGC-7901,人非小细胞肺癌细胞株A549和人成纤维肉瘤细胞株HT-1080的体外细胞毒活性.结果显示:化合物AA-Ⅲ2的IC50值较小,对上述3种细胞有较好的抑制作用,值得进一步研究.
积雪草酸; 五环三萜化合物; 合成; 抗肿瘤活性
积雪草酸(2α,3β,23-三羟基-乌苏烷-12-烯-28-酸),又名亚细亚酸(asiatic acid,AA),属于乌苏烷型骨架的五环三萜类化合物,具有广泛的药理活性,包括抗肿瘤[1-2]、神经保护作用[3-4]、保护肝脏和肾脏[5-8]、抗炎[9]以及影响血管生成[10-11]等生物活性.因此,积雪草酸具有广泛的开发前景.为了深入研究积雪草酸的结构与其抗肿瘤活性的关系,本文以积雪草酸为先导化合物,对其C-2、C-3、C-23位羟基和C-28位羧基等官能团进行结构修饰,设计并合成了3类共10个化合物,所合成化合物均未见文献报道.采用MTT法,对部分积雪草酸衍生物进行体外细胞毒活性的药理评价,为进一步研究积雪草酸的构效关系研究奠定了基础.3条合成路线如下所示:
路线1 2-肟基-3β,23-二羟基-乌苏烷-12-烯-28-羧酸(酯、胺)类化合物合成
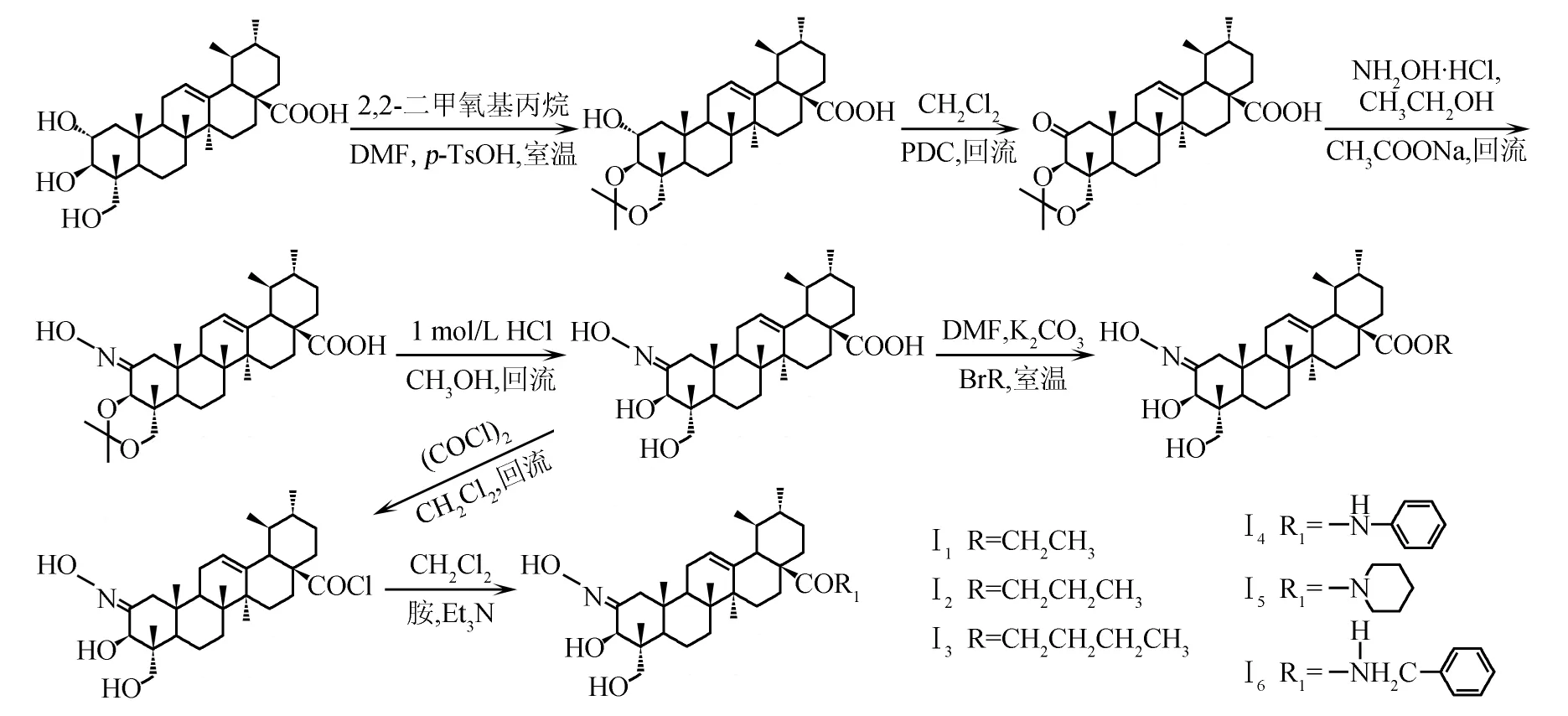
路线2N-[2-肟基-3β,23-O-异亚丙基-乌苏烷-12-烯-28-酰]-氨基醇类化合物的合成
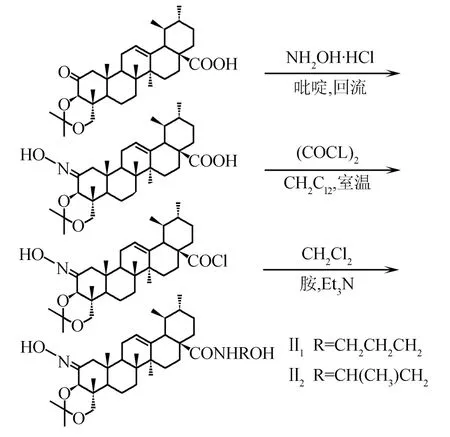
路线3N-[1,2-骈噁唑-3β,23-二羟基-乌苏烷-12-烯-28-酰]-氨基醇类化合物的合成
1 实验部分
1.1 仪器与试剂
实验所用试剂均为分析纯或化学纯,使用时根据需要进一步纯化.反应所用溶剂按常规方法加以精制.以薄层色谱(TLC)检测反应进程,薄层色谱用高效色谱硅胶GF254,柱层析色谱用200~300目柱色谱硅胶.在Buchi B-545熔点仪上测定化合物熔点,温度未经校正.在Thermo Nicolet 470FT红外光谱仪上测定(KBr压片)红外光谱.在菲尼根hermo-finnigan LCQ equipment仪上测定质谱.在Bruker ARX-500型核磁共振分析仪上测定核磁共振氢谱,CDCl3为溶剂,TMS为内标.
1.2 中间体的合成
1.2.1 2-肟基-3β,23-二羟基-乌苏烷-12-烯-28-羧酸的制备(AA-Ⅰ)
将AA(200 mg,0.40 mmol)和对甲基苯磺酸(34 mg,0.20 mmol),溶解在DMF(N,N-二甲基甲酰胺)中,然后滴加2,2-二甲氧基丙烷(1.0 mL,4.0 mmol),在室温下搅拌,反应完毕,用质量分数5 %的氢氧化钠溶液调节pH至7~8,混合物用乙酸乙酯提取,有机层用蒸馏水及饱和氯化钠溶液洗涤,无水MgSO4干燥,抽滤,减压蒸馏得白色固体AA-1.m.p.156.4~157.8 ℃.IR(KBr),σ/cm-1:3 400,1 698,1 200 cm-1.
再将硅胶负载PDC(重铬酸吡啶钅翁,160 mg)加入到二氯甲烷中,加入0.2 mL乙酸酐搅拌30 min,而后将此溶液加到化合物AA-1的二氯甲烷溶液中,回流2 h.反应结束后,过滤除去硅胶,滤液减压蒸出溶剂,再溶于30 mL乙酸乙酯,搅拌30 min,用水萃取,有机相用无水Na2SO4干燥,抽滤除去干燥剂,旋蒸得到粗品.粗品经柱层析提纯后得化合物AA-2.1H NMR(500 MHz,CDCl3),δ:5.24(s,1H,H-12),3.54(d,1H,J=11.2 Hz,H-23),3.50(s,1H,H-3),3.48(d,1H,J=11.2Hz,H-23),2.40(s,1H,H-9),2.17(d,1H,J=11.2,H-18),1.25,1.15,1.13,1.04,0.98,0.75(s,each 3H),0.90(d,3H,J=6.0 Hz),0.86(d,3H,J=6.0 Hz).ESI-MS,m/z:527.4(M+H)+.
将化合物AA-2用无水乙醇溶解,按摩尔比1∶1∶1.2的比例分别加入AA-2、乙酸钠和盐酸羟胺,在78 ℃下回流,TLC检测反应终点.然后蒸干溶剂,充分水洗,过滤,滤饼洗至中性,再用甲醇溶解,并加入1 mol/L的盐酸数滴,在室温下搅拌3 h,反应结束后,蒸除溶剂,剩余混合物用乙酸乙酯提取,有机层用无水MgSO4干燥,抽滤,减压蒸馏得白色固体,粗品用硅胶柱色谱纯化,得产物AA-Ⅰ(36 mg).IR(KBr),σ/cm-1:1 379,1 696,2 925,3 422.1H NMR(500 MHz,CDCl3),δ:5.27(s,1H,H-12),3.52(d,1H,J=11.2 Hz,H-23),3.50(s,1H,H-3),3.48(d,1H,J=11.2 Hz,H-23),2.43(s,1H,H-9),2.14(d,1H,J=10.8 Hz,H-18),1.33,1.25,1.12,1.09,1.04,0.80(s,each 3H).ESI-MS,m/z:502.4(M+H)+.
1.2.2 2-肟基-3β,23-O-异亚丙基-乌苏烷-12-烯-28-羧酸(AA-Ⅱ)的制备
将化合物AA-2用适量吡啶溶解,按摩尔比1∶2的比例分别加入化合物AA-2和盐酸羟胺,115 ℃下回流,TLC检测.反应结束后,冷却至室温,再加入冰水抽滤,水洗滤饼至中性,经柱层析分离后得固体AA-Ⅱ(48 mg).IR(KBr),σ/cm-1:1 049,1 275,1 695,2 924,3 419.1H NMR(500 MHz,CDCl3),δ:5.27(s,1H,H-12),3.54(d,1H,J=10.8 Hz,H-23),3.51(s,1H,H-3),3.46(d,1H,J=10.8 Hz,H-23),2.42(s,1H,H-9),2.14(d,1H,J=10.6 Hz,H-18),1.35,1.33,1.13,1.06,1.04,0.98,(s,each 3H),0.92(d,3H,J=6.0 Hz),0.87(d,3H,J=6.0 Hz).ESI-MS,m/z:542.4(M+H)+.
1.2.3 1,2-骈噁唑-3β,23-二羟基-乌苏烷-12-烯-28-羧酸(AA-Ⅲ)的制备
将化合物AA-2用甲酸乙酯溶解,加入少量甲醇钠,室温下反应24 h,用TLC跟踪检测确定反应终点.减压蒸除溶剂,用乙酸乙酯和水萃取,有机相用无水Na2SO4干燥,抽滤除去干燥剂,旋蒸.将粗品溶解在4 mL二氯甲烷中,加入少量盐酸羟胺,室温搅拌,用TLC跟踪检测确定反应终点.混合物用乙酸乙酯提取,有机层用蒸馏水及饱和NaCl溶液洗涤,无水MgSO4干燥,抽滤,减压蒸馏得白色固体.然后将固体溶解在甲醇中,加入1 mol/L盐酸数滴,搅拌3 h.减压蒸除溶剂,用乙酸乙酯和水萃取,旋蒸后的粗品经柱层析得产物AA-Ⅲ.IR(KBr),σ/cm-1:525,1 255,2 492,2 678,2 739cm-1.1H NMR(500 MHz,CDCl3),δ:5.26(s,1H,H-12),3.62(d,1H,J=10.6 Hz,H-23),3.60(s,1H,H-3),3.46(d,1H,J=10.6 Hz,H-23),2.40(s,1H,H-9),2.20(d,1H,J=11.2 Hz,H-18),2.04(s,1H),1.29,1.16,1.14,1.09,0.96,0.88(s,each 3H).ESI-MS,m/z:512.3(M+H)+.
1.3 目标化合物的合成
1.3.1 2-肟基-3,23-二羟基-乌苏烷型-12-烯-28-羧酸酯类化合物(AAI1~I3)的制备
1.3.1.1 2-肟基-3β,23-二羟基-乌苏烷-12-烯-28-乙酯(AA-Ⅰ1)的制备
将化合物AA-Ⅰ(50 mg,0.099 8 mmol)溶解在4 mL DMF中,加入催化剂量的碳酸钾,再滴入溴乙烷(0.028 mL,0.380 mmol),室温搅拌,用TLC跟踪检测确定反应终点.混合物用乙酸乙酯提取,有机层用蒸馏水及饱和氯化钠溶液洗涤,无水MgSO4干燥,抽滤,减压蒸馏得白色固体,粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=6/1,得白色粉末状固体AA-Ⅰ1(22 mg).m.p.210.7~211.6 ℃.IR(KBr),σ/cm-1:1 040,1 453,1 667,2 926,3 425.1H NMR(500 MHz,CDCl3),δ:5.27(s,1H,H-12),4.30(d,1H,J=7.0 Hz,H-23),4.06(d,1H,J=7.0 Hz,H-23),3.61~3.67(m,2H,OCH2),3.50(s,1H,H-3),2.00(s,1H,NOH),1.42~1.43(t,3H,CH3),1.26,1.11,0.95,0.87,0.76,0.69(s,each 3H).ESI-MS,m/z:530.5(M+H)+.
1.3.1.2 2-肟基-3β,23-二羟基-乌苏烷-12-烯-28-丙酯(AA-Ⅰ2)的制备
按照化合物AA-Ⅰ1的合成方法,由化合物AA-Ⅰ和溴代正丙烷反应.粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=6/1,得白色粉末状固体AA-Ⅰ2(18 mg).IR(KBr),σ/cm-1:1 052,1 454,1 662,2 927,3 417.m.p.213.2~214.1 ℃.1H NMR(500 MHz,CDCl3),δ:5.27(s,1H,H-12),4.28~4.32(t,2H,OCH2—),4.08(d,1H,J=8.5 Hz,H-23),3.94(d,1H,J=8.5Hz,H-23),3.33(s,1H,H-3),2.38(s,1H,H-9),2.26(d,1H,J=9.0 Hz,H-18),2.00(s,1H,NOH),1.25,1.11,0.94,0.76(s,each 3H),1.61~1.63(m,2H,—CH2—),0.95~0.96(t,3H,—CH3).ESI-MS,m/z:544.3(M+H)+.
1.3.1.3 2-肟基-3β,23-二羟基-乌苏烷-12-烯-28-丁酯(AA-Ⅰ3)的制备
按照化合物AA-Ⅰ1的合成方法,由化合物AA-Ⅰ和溴代正丁烷反应.粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=6/1,得白色粉末状固体AA-Ⅰ3(16 mg).IR(KBr),σ/cm-1:1 046,1 454,1 661,2 927,3 420.m.p.222.5~224.3 ℃.1H NMR(500 MHz,CDCl3),δ:5.27(s,1H,H-12),4.08~4.16(t,2H,OCH2—),3.84(d,1H,J=10.0 Hz,H-23),3.58(d,1H,J=10.0 Hz,H-23),3.35(s,1H,H-3),2.43(d,1H,J=10.0 Hz,H-18),2.36(s,1H,H-9),1.74~1.76(m,2H,—CH2a—),1.48~1.51(m,2H,—CH2b—),1.34,1.23,1.21,0.91,0.87,0.85(s,each 3H),0.96~0.98(t,3H,—CH3).ESI-MS,m/z:573.7(M+NH3)+.
1.3.2N-2-肟基-3β,23-二羟基-乌苏烷-12-烯-28-酰]-胺类化合物(AAI4~I6)的制备
1.3.2.1N-[2-肟基-3β,23-二羟基-乌苏烷-12-烯-28-酰]-苯胺(AA-Ⅰ4)的制备
将化合物AA-Ⅰ(50 mg,0.099 8 mmol)溶解在4 mL二氯甲烷中,加入草酰氯((COCl2),0.4 mmol),室温搅拌20 h,生成2-肟基-3β,23-二羟基-乌苏烷-12-烯-28-酰氯,蒸除反应溶剂和未反应的草酰氯,残余物加入2 mL环己烷,减压蒸除环己烷,反复操作2次.酰氯中加入2 mL二氯甲烷使之完全溶解后,加三乙胺调节pH为9~10,搅拌5 min后,加入苯胺(32 mg,0.4 mmol),室温下反应,TLC监测反应终点.反应结束后,减压蒸除二氯甲烷,向反应液中加入2 mL水,以2 mol/L盐酸调pH至3~4,析出黄色固体,迅速减压抽滤,水洗滤饼至中性.粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=4/1,得化合物AA-Ⅰ4(15 mg).m.p.184.0~184.9 ℃.IR(KBr),σ/cm-1:1 502,1 388,1 678,2 926,3 417.1H NMR(500 MHz,CDCl3),δ:7.43(d,2H,2CHa),7.19(d,1H,CHb),7.23(s,1H,NH),5.27(s,1H,H-12),3.76(d,1H,J=7.0 Hz,H-23),3.21(s,1H,H-3),3.40(d,1H,J=7.0 Hz,H-23),2.40(s,1H,H-9),2.17(d,1H,J=11.2 Hz,H-18),1.25,1.15,1.13,1.04,0.98,0.75(s,each 3H).ESI-MS,m/z:574.0(M+H)+.
1.3.2.2N-[2-肟基-3β,23-二羟基-乌苏烷-12-烯-28-酰]-环己胺(AA-Ⅰ5)的制备
按照化合物AA-Ⅰ4的合成方法,由化合物AA-Ⅰ和环己胺制备而成.粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=4/1,得黄色固体AA-Ⅰ5(13 mg).m.p.182.5~183.4 ℃.IR(KBr),σ/cm-1:602,1 040,1 637,2 924,3 445.1H NMR(500 MHz,CDCl3),δ:5.27(s,1H,H-12),3.76(d,1H,J=7.0 Hz,H-23),3.14(s,1H,H-3),3.40(d,1H,J=7.0 Hz,H-23),2.40(s,1H,H-9),2.17(d,1H,J=11.2 Hz,H-18),1.25,1.18,1.13,0.96,0.89,0.88(s,each 3H).ESI-MS,m/z:568.8(M+H)+.
1.3.2.3N-[2-肟基-3β,23-二羟基-乌苏烷-12-烯-28-酰]-苄胺(AA-Ⅰ6)的制备
按照化合物AA-Ⅰ4的合成方法,由化合物AA-Ⅰ和苄胺制备而成.粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=3/1,得黄色固体AA-Ⅰ6(14 mg).m.p.185.7~186.8 ℃.IR(KBr),σ/cm-1:1 025,1 454,1 651,2 927,3 282.1H NMR(500 MHz,CDCl3),δ:7.33(m,2H,2CHb),7.23~7.26(m,3H,CHa),5.27(s,1H,H-12),3.54(d,1H,J=11.2 Hz,H-23),3.50(s,1H,H-3),3.48(d,1H,J=11.2 Hz,H-23),2.40(s,1H,H-9),2.17(d,1H,J=11.2 Hz,H-18),1.32,1.14,1.11,1.04,0.99,0.84(s,each 3H).ESI-MS,m/z:591.4(M+H)+.
1.3.3 2-肟基-3β,23-O-异亚丙基-乌苏烷-12-烯-28-氨基醇类化合物的(AAⅡ1~Ⅱ2)制备
1.3.3.1N-[2-肟基-3β,23-O-异亚丙基-乌苏烷-12-烯-28-酰]-3-氨基-1-丙醇(AA-Ⅱ1)的制备
将化合物AA-Ⅱ(50 mg,0.092 4 mmol)溶解在4 mL二氯甲烷中,加入草酰氯(0.4 mmol),室温搅拌20 h,生成2-肟基-3β,23-O-异亚丙基-乌苏烷-12-烯-28-酰氯,蒸除反应溶剂和未反应的草酰氯,残余物加入2 mL环己烷,减压蒸除环己烷,反复操作2次.酰氯中加入2 mL二氯甲烷使之完全溶解后,加三乙胺调pH为9~10,搅拌5 min后,加入3-氨基-1-丙醇(30 mg,0.4 mmol),室温下反应,TLC监测反应终点.反应结束后,减压蒸除二氯甲烷,向反应液中加入2 mL水,以2 mol/L盐酸调pH至3~4,析出白色固体,迅速减压抽滤,水洗滤饼至中性.室温干燥得粗品,经硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=4/1,得白色粉末状固体AA-Ⅱ1(19 mg).m.p.234.3~235.2 ℃.IR(KBr),σ/cm-1:1 064,1 655,1 726,2 937,3 420.1H NMR(500 MHz,CDCl3),δ:8.04(s,1H,NH),5.27(s,1H,H-12),3.83~3.86(m,1H,NCH),3.77(d,1H,J=9.6 Hz,H-23),3.50(s,1H,H-3),3.34(d,1H,J=9.6 Hz,H-23),3.25(d,2H,J=9.2 Hz,CH2O),2.23(d,1H,J=11.0 Hz,H-18),1.32(d,3H,J=8.4 Hz,CH3),1.25,1.15,1.13,1.04,0.98,0.75(s,each 3H),0.92(d,3H,J=6.4 Hz),0.87(d,3H,J=6.4 Hz),0.92(d,3H,J=6.0 Hz),0.87(d,3H,J=6.0 Hz).ESI-MS,m/z:598.4(M+H)+.
1.3.3.2N-[2-肟基-3β,23-O-异亚丙基-乌苏烷-12-烯-28-酰]-2-氨基-2-甲基乙醇(AA-Ⅱ2)的制备
按照化合物AA-Ⅱ1的合成方法,由化合物AA-Ⅱ和2-氨基-1-丙醇制备而成.粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=3/1,得白色固体AA-Ⅱ2(18 mg).m.p.230.5~232.1 ℃.IR(KBr),σ/cm-1:1 064,1 655,1 726,2 937,3 420.1H NMR(500 MHz,CDCl3),δ:8.03(s,1H,NH),5.27(s,1H,H-12),3.24~3.26(t,1H,NCH2),3.77(d,1H,J=9.6 Hz,H-23),3.50(s,1H,H-3),3.34(d,1H,J=9.6 Hz,H-23),3.50(t,2H,CH2O),3.25(d,2H,J=9.2 Hz,CH2O),2.23(d,1H,J=11.0 Hz,H-18),1.58(m,2H,—CH2—),1.22,1.15,1.11,1.04,0.98,0.78(s,each 3H),1.14(d,3H,J=5.7 Hz),0.92(d,3H,J=6.3 Hz).ESI-MS,m/z:598.3(M+H)+.
1.3.4N-[1,2-骈噁唑-3β,23-二羟基-乌苏烷-12-烯-28-酰]-氨基醇乙酸酯类化合物(AAⅢ1~Ⅲ2)的制备
1.3.4.1N-[1,2-骈噁唑-3β,23-二羟基-乌苏烷-12-烯-28-酰]-3-氨基-1-丙醇乙酸酯(AA-Ⅲ1)的制备
将化合物AA-Ⅲ(100 mg,0.092 4 mmol)溶解在4 mL二氯甲烷中,加入草酰氯(0.4 mmol),室温搅拌20 h,蒸除反应溶剂和未反应的草酰氯,残余物加入2 mL环己烷,减压蒸除环己烷,反复操作2次.酰氯中加入2 mL二氯甲烷使之完全溶解后,加三乙胺调pH为9~10,搅拌5 min后,加入3-氨基-1-丙醇(30 mg,0.4 mol/L),室温下反应,TLC监测反应终点.反应结束后,减压蒸除二氯甲烷,向反应液中加入2 mL水,以2 mol/L盐酸调pH至3~4,析出淡黄色固体,迅速减压抽滤,水洗滤饼至中性.室温干燥得粗品淡黄色固体AA-4.
将经过硅胶柱提纯的固体AA-4溶于二氯甲烷中,加入乙酸酐及少量DMAP(二甲氨基吡啶),室温下搅拌,反应完毕,蒸出溶剂,加入3 mL水分散固体,以2 mol/L盐酸调pH至3~4,抽滤,水洗至中性,室温干燥得淡黄色粉末状固体AA-Ⅲ1(13 mg).m.p.213.6~214.7 ℃.IR(KBr),σ/cm-1:1 036,1 724,2 928,3 872.1H NMR(500 MHz,CDCl3),δ:8.11(s,1H,NH),7.83(s,1H,N==CH),5.27(s,1H,H-12),3.54(d,1H,J=11.2 Hz,H-23),3.50(s,1H,H-3),3.48(d,1H,J=11.2 Hz,H-23),3.12~3.20(t,2H,NCH2),2.40(s,1H,H-9),2.22(s,3H,CH3),2.17(d,1H,J=11.2 Hz,H-18),1.25,1.15,1.13,1.04,0.98,0.75(s,each 3H).ESI-MS,m/z:629.0(M+H2O)+.
1.3.4.2N-[1,2-骈噁唑-3β,23-二羟基-乌苏烷-12-烯-28-酰]-2-氨基-2-甲基乙醇乙酸酯(AA-Ⅲ2)的制备
按照化合物AA-Ⅲ1的合成方法,由化合物AA-Ⅲ和2-氨基-1-丙醇制备而成.粗品用硅胶柱色谱纯化,洗脱剂为V(石油醚)/V(乙酸乙酯)=3/1,得淡黄色粉末状固体AA-Ⅲ2(14 mg).m.p.214.1~214.9 ℃.IR(KBr),σ/cm-1:1 140,1 455,1 758,2 928,3 422 .1H NMR(500 MHz,CDCl3),δ:7.83(s,1H,N==CH),5.27(s,1H,H-12),4.49~4.51(t,2H,CH2O),3.52(d,1H,J=11.2 Hz,H-23),3.50(s,1H,H-3),3.44(d,1H,J=11.2 Hz,H-23),2.40(s,1H,H-9),2.24(s,3H,CH3),2.17(d,1H,J=11.2 Hz,H-18),1.25,1.15,1.13,1.04,0.98,0.75(s,each 3H).ESI-MS,m/z:629.4(M+H2O)+.
1.4 初步体外细胞毒活性测试
选用人胃腺癌细胞株SGC-7901,人非小细胞肺癌细胞株A549和人成纤维肉瘤细胞株HT-1080为靶细胞,采用四唑盐比色法即MTT法,对化合物AA、AA-Ⅰ1、AA-Ⅰ5和AA-Ⅲ2进行体外细胞毒活性测试.
1.4.1 埋板
将生长至对数生长期的细胞消化、离心,制成细胞悬液,吹匀并将SGC-7901、A549、HT-1080三种细胞其密度分别调至2×104个/mL、2.5×104个/mL、1×104个/mL,以180 μL/孔接种到96孔板中,加的过程中不断吹匀细胞,保证细胞密度一致,铺板过程要迅速,保证每孔的细胞数目大致相同,将96孔板置于37 ℃、含5 %的CO2、饱和湿度的培养箱中孵育24 h.
1.4.2 加药
将药物配制成以完全培养基为溶剂,初始质量浓度为1 g/L的溶液,将上述溶液稀释成5个剂量组,药物的质量浓度分别为500,50, 5,0.5,0.05 mg/L.取各个剂量的药液加入96孔板中,每孔20 μL,每剂量3个复孔.加药后的药物终浓度分别为50,5,0.5,0.05,0.005 mg/L.加药完毕放入培养箱中孵育72 h.
1.4.3 MTT检测
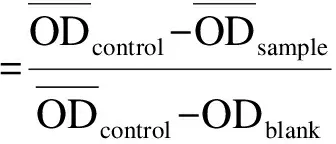
将加药并于培养箱中孵育48 h后的96孔板中培养基轻轻甩出,然后加入配置好的MTT溶液(5 g/L),每孔20 μL,放入培养箱中继续孵育3 h.取出孵育3 h的96孔板,将其中的MTT溶液轻轻甩出,每孔加100 μL的DMSO,微量振荡仪上振荡5 min以使甲瓒完全溶解,用酶标仪在490 nm下测光密度(OD)值,并记录数据.按下列公式计算药物对肿瘤细胞体外增殖的抑制率(Inhibition Rate,IR):

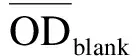
用IC50计算软件来计算半数抑制浓度(IC50).
2 结果与讨论
2.1 化学合成
以积雪草酸为起始原料,合成了2-肟基-3β,23-二羟基-乌苏烷-12-烯-28-羧酸(酯、胺)类、N-[2-肟基-3β,23-O-异亚丙基-乌苏烷-12-烯-28-酰]-氨基醇类和N-[1,2-骈噁唑-3β,23-二羟基-乌苏烷-12-烯-28-酰]-氨基醇乙酸酯类3类共10个化合物,均未见文献报导,其结构通过IR、MS和1H NHR得以确认.
2.2 生物活性评价
采用四唑盐比色法即MTT法对化合物的抗肿瘤活性进行筛选.实验结果见表1和表2.由表1、表2数据可知:积雪草酸衍生物对三种肿瘤细胞有一定抑制作用.其中化合物AA-Ⅲ2在50 mg/L质量浓度中,对于SGC-7901、HT-1080、A549的抑制率分别为93.06 %,96.85 %,95.52 %,明显优于对照品吉非替尼(73.25 %,85.01 %,68.15 %),与阿霉素的抑制活性基本相同,甚至对于A549肿瘤细胞抑制作用要强于阿霉素(90.25 %).而化合物AA-Ⅰ1,AA-Ⅰ5在50 mg/L质量浓度中,对于SGC-7901、HT-1080、A549的抑制率相对较低,具有较弱的抑制活性.化合物AA-Ⅲ2(IC50:12.395 μmol/L,13.293 μmol/L)对SGC-7901、HT-1080的细胞毒性与吉非替尼(10.527 μmol/L,12.998 μmol/L)相当,其中对A549(8.052 μmol/L)的细胞毒性明显优于吉非替尼(23.496 μmol/L),并且AA-Ⅲ2和AA相比,对于A549、HT-1080两种肿瘤细胞有着明显的优势.
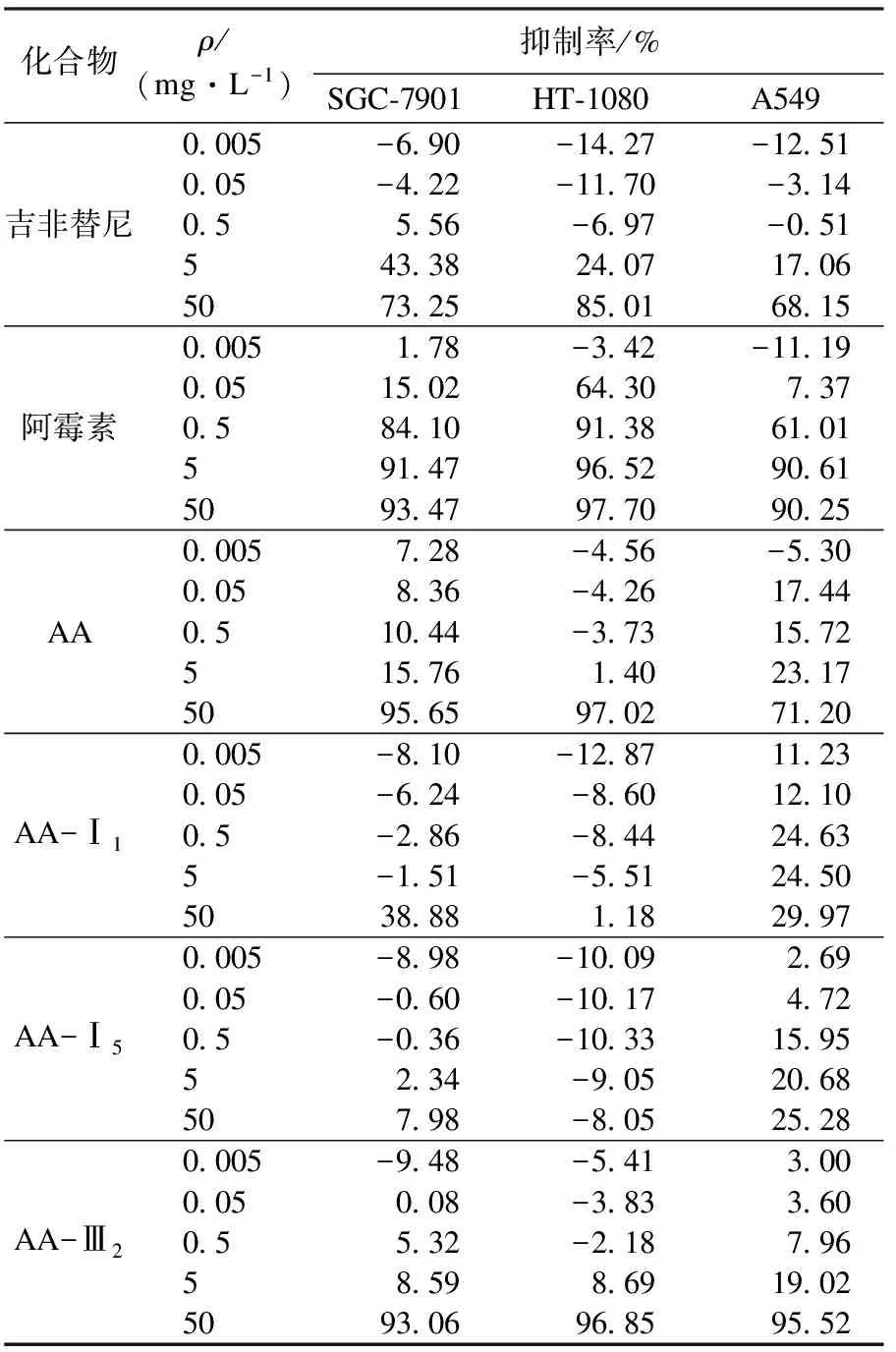
表1 化合物在不同质量浓度下对三种细胞株的抑制率

表2 化合物的IC50值
3 结 论
初步的构效关系表明:28位成氨基醇乙酸酯对SGC-7901、A549、HT-1080三种肿瘤细胞的抗肿瘤活性明显增强,如AA-Ⅲ2≈阿霉素>>吉非替尼,甚至对于A549抑制作用要略高于阿霉素;28位成酯的衍生物,对SGC-7901、A549、HT-1080三种肿瘤细胞的抗肿瘤活性要高于28位成酰胺的衍生物,如AA-Ⅰ1>AA-Ⅰ5;1位成肟可能对积雪草酸对SGC-7901、A549、HT-1080三种肿瘤细胞的抗肿瘤活性起到了抑制作用,如AA>> AA-Ⅰ1>AA-Ⅰ5,而1,2位骈成噁唑环可能对积雪草酸的抗肿瘤尤其是对A549细胞起到了促进作用,如AA-Ⅲ2>>AA.
[1] 张海峰.积雪草酸抗肿瘤作用的初步研究[D].镇江:江苏大学药学院,2008:14-18.
[2] 吕婷婷,陈燕,姜旭东,等.积雪草酸对慢性髓细胞白血病急变株K562细胞增殖的影响及其机制[J].中国医院药学杂志,2010,30(17):1428-1431.
[3] 高静,熊御云,徐敏芳,等.积雪草酸对抗神经损伤的作用与保护线粒体有关[G]//高静.2011全国老年痴呆与衰老相关疾病学术会议第三届山东省神经内科医师(学术)论坛论文汇编.北京:中国药理学会,2011:20
[4] Krishnamurthy R G,Senut M C,Zemke D,et al.Asiatic Acid,a Pentacyclic Triterpene from Centella Asiatica,is Neuroprotective in a Mouse Model of Focal Cerebral Ischemia[J].Journal of Neurosci-ence Research,2009,87(11):2541-2550.
[5] 陈杰,曾令兰,王华,等.积雪草酸对HSC-T6细胞Smad7和PPARγ的mRNA表达的影响[J].临床肝胆病杂志,2011,27(3):251-253.
[6] Guo W J,Liu W,Hong S C,et al.Mitochondria-Dependent Apoptosis of Con A-Activated T Lymphocytes Induced by Asiatic Acid for Preventing Murine Fulminant Hepatitis[J].PLoS One,2012,7(9):e46018.
[7] 李文,吴晨光,王丽,等.积雪草酸对高糖培养大鼠肾小球系膜细胞氧化应激及细胞外基质分泌的影响[J].江苏医药,2011,37(21):2484-2486.
[8] 陈宇宁,陈艳,王丽,等.积雪草酸对糖尿病大鼠肾脏c-Jun氨基末端激酶信号通路的影响[J].南京医科大学学报:自然科学版,2011,31(3):399-402.
[9] Fan Y M, Xu L Z, Gao J,et al.Phytochemical and Antiinflammatory Studies onTerminaliaCatappa[J].Fitoterapia,2004,75(3/4):253-260.
[10]Kavitha C V,Agarwal C,Agarwal R,et al.Asiatic Acid Inhibits Pro-Angiogenic Effects of VEGF and Human Gliomas in Endothelial Cell Culture Models[J].PLoS One,2011,6(8):e22745.
[11]姜旭东.积雪草酸对血管生成的影响及其机制初探[D].武汉:华中科技大学,2011:13-19.
Synthesis and Characterization of Asiatic Acid Derivatives and Its Anti-tumor Activities
WANG Nan, ZHANG Meng, MENG Yan-qiu
(Shenyang University of Chemical Technology, Shenyang 110142, China)
Natural active product is an important source of anti-cancer drugs.Through C-2,C-3,C-23 and C-28 for structural modification,three types of ten new compounds of asiatic acid derivatives were designed and synthesized.The target compounds were not reported,and confirmed its structure by IR,MS,1H NMR.Anti-tumor activities of asiatic acid and these derivatives against SGC-7901,A549 and HT-1080 cells in vitro were investigated by MTT assay.The results indicated that compound AA-III2had stronger cell growth inhibitor than other products.It is worth to be intensively further studies.
asiatic acid; pentacyclic triterpenoid; synthesis; anti-tumor activity
2014-02-27
王楠(1987-),女,辽宁沈阳人,硕士研究生在读,国家奖学金获得者,主要从事五环三萜类天然产物结构改造及其抗癌活性的研究.
孟艳秋(1963-),女,辽宁义县人,教授,博士,主要从事天然化合物有效成分结构改造及抗癌活性和新型半合成抗生素方面的研究.
2095-2198(2015)03-0242-07
10.3969/j.issn.2095-2198.2015.03.011
R914.5
A
猜你喜欢
福建文学(2019年12期)2019-08-06
炎黄地理(2017年10期)2018-01-31
少年文艺·开心阅读作文(2017年1期)2017-02-24
材料科学与工程学报(2016年4期)2017-01-15
高原山地气象研究(2016年1期)2016-11-10
饮食科学(2016年3期)2016-07-04
饮食科学(2016年3期)2016-07-04
食品工业科技(2014年13期)2014-03-11
