
立方相Ag3PO4(111)面原子几何及弛豫结构的第一性原理计算
2015-03-22 10:28马新国徐国旺黄楚云
原子与分子物理学报 2015年6期
马新国, 刘 娜, 祝 林, 徐国旺,2, 黄楚云,2
(1. 湖北工业大学理学院, 武汉430068; 2. 湖北工业大学太阳能高效利用湖北省协同创新中心, 武汉 430068)
立方相Ag3PO4(111)面原子几何及弛豫结构的第一性原理计算
马新国1, 2, 刘 娜1, 祝 林1, 徐国旺1,2, 黄楚云1,2
(1. 湖北工业大学理学院, 武汉430068; 2. 湖北工业大学太阳能高效利用湖北省协同创新中心, 武汉 430068)
采用平面波超软赝势方法研究了立方相Ag3PO4(111)面的表面能和表面原子弛豫结构. 首先对Ag3PO4(111)面的八种不同原子终止结构的体系总能量进行计算, 结果表明B种表面模型被证实为最稳定的(111)面原子几何结构. 针对该表面结构, 探讨了表面能和原子弛豫与模型中原子层数和真空厚度的关系, 当原子层数为24层, 真空厚度为0.6 nm时, 表面能收敛于1.41 J/m2(LDA-CAPZ)和1.39 J/m2(GGA-PBE). 表面原子弛豫后, 表面两个三配位的Ag原子均向里移动, 超过0.06 nm, 而表面次层的O原子则均向外移动约0.0042 nm, 导致弛豫后暴露在最表面的是O原子, 同时表面原子的核外电子向表面内部发生转移, 结构趋于稳定. 这些结果为进一步深入研究Ag3PO4表面的光催化活性起源提供理论支持.
第一性原理; 磷酸银; 表面结构; 弛豫
1 引 言
寻找可见光辐照下可直接光解水制氢和光降解污染物的高活性半导体光催化材料一直是光催化领域的研究热点[1, 2]. 目前具有比P25(TiO2)性能更优越的金属氧化物光催化材料屈指可数[3]. 2010年,朱永法等首次发现单斜相BiPO4具有极高的光催化活性, 其面积比活性是P25(TiO2)的30倍以上, 但是其对光谱的响应小于300 nm[4, 5]. 随后, 叶金花等[6]发现立方相Ag3PO4的一种新用途: 以磷酸银为催化剂, 降解亚甲基蓝过程中其催化速率比单斜相BiVO4和商业TiO2-xNx光催化剂要快数十倍, 其吸收阀值大于500 nm, 是一种极具发展潜力的新型可见光催化材料. Ag3PO4半导体材料具有强氧化性的特点已经引起了大家的广泛关注. 目前的研究主要集中在材料的化学合成与微观形貌分析[6-8], 在晶体结构与光催化性能之间的关系等方面已经有相关报道[9,10].
光催化剂的表面结构很大程度决定着其表面光催化活性和反应过程. 一般而言, 具有高表面能的晶面具有更高的光催化活性. 然而, 在化学合成过程中, 具有高活性的晶面往往很小或者很快消失. 为此增大高活性晶面的表面积, 需要特定的合成条件. 例如TiO2(101)面的表面能为0.44 J/m2, 其活性较低, 而(001)的表面能为0.9 J/m2, 却显示出了更高的活性[11]. 最近,Ye等[8]开展了Ag3PO4晶面与光催化性能的系统研究, 他们通过控制特定晶面的生长, 有效提高了Ag3PO4的光催化活性. 进一步, 他们成功合成出了具有Ag3PO4(111)面的半导体光催化剂, 暴露的(111)面具有比(100)和(110)面更高的表面能, 因而显示出了更高的光氧化活性, 即(111)面具有10倍于(100)和(110)面的氧气生成率[12]. Zheng等[13]也对这些表面的光催化活性进行了深入研究, 得出了相似结果. 尽管实验已经证实了Ag3PO4(111)面具有最高的光催化活性, 但是在实验上精确的探测Ag3PO4(111)面完整的表面结构信息仍然十分困难, 致使目前在材料的表面原子几何结构与光催化活性之间的机理研究仍然没有完全展开. 因此, 本文试图建立Ag3PO4(111)面模型, 通过计算表面模型的体系总能量和原子弛豫过程, 确定表面的原子几何结构, 为进一步深入理解该表面的活性起源提供理论支持.
2 物理模型和计算方法
立方相Ag3PO4晶体结构的空间群为P4-3n(NO. 218). 采用XRD(Cu Kα)获得的晶体结构以PO4四方体为单元构成的体心立方结构, 即PO4单元位于八个顶点和体心位置上[14]. 尤其需要注意的是晶胞中六个Ag原子被分配到十二个位置上, 也就是说, 这十二个位置中有一半位置被Ag占据, 有一半为空位置, 这里Ag原子坐标为(0.25, 0, 0.50). 图1显示了Ag原子与周围的四个O原子配位, P原子与周围的四个O原子配位. 而每个O原子与周围的三个Ag原子和一个P原子形成四配位. 对该结构沿垂直[111]方向进行剪切, 由于断键情况和原子位置的不同, 存在八种不同的表面原子终止结构, A类结构: 最表层终止于三个Ag原子, 均为三配位, 次层是三个O原子, 均为三配位, 第三层是O原子, 为三配位. B类结构: 最表层终止于两个Ag原子, 均为三配位, 次层是三个O原子, 其中两个为三配位, 一个为二配位, 第三层是O原子, 为三配位. C类结构: 最表层终止于一个Ag原子, 为三配位, 次层是三个O原子, 其中一个为三配位, 两个为二配位, 第三层是O原子, 为三配位. D类结构: 最表层终止于三个O原子, 均为二配位, 次层是O原子, 为三配位. E类结构: 最表层终止于两个O原子, 均为二配位, 次层是O原子, 为三配位, 第三层是P原子, 为三配位. F类结构: 最表层终止于一个O原子, 为二配位,次层是O原子, 为三配位, 第三层是P原子, 为二配位. G类结构: 最表层终止于一个O原子, 为三配位,次层是P原子, 为一配位. H类结构: 最表层终止于一个P原子, 为一配位, 次层是Ag原子, 均为三配位. 其剪切位置如图1所示.
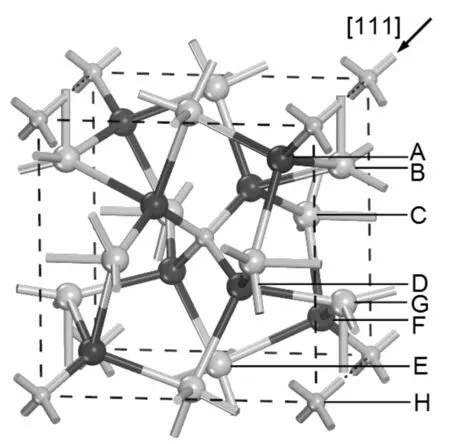
图1 立方相Ag3PO4的晶胞结构及垂直[111]方向的八种剪切位置. 黑色球、小灰球、大灰球分别表示O、P和Ag原子Fig. 1 The crystal cell of cubic Ag3PO4 and the eight sliced sites at vertical [111] direction. Black, small gray and big gray spheres represent the O, P and Ag atoms, respectively
本工作采用了基于密度泛函理论的平面波超软赝势方法研究Ag3PO4半导体表面的原子几何结构和弛豫结构[15]. 在计算八种不同的表面原子终止结构的体系能量时, 分别采用了局域密度近似(LDA)中的CA-PZ方案[16]和广义梯度近似(GGA)中的PBE方案[17], 在研究表面原子弛豫时采用了PBE方案. 在描述离子实与价电子之间的相互作用时, 选取的价电子组态分别为O: 2s22p4,P: 3s23p3,Ag: 4d105s1. 平面波截断能设置为400 eV, 第一布里渊区采用Monkhorst的K空间网格6×6×1[18]. 在几何优化计算中, 自恰收敛精度设置为1×10-6eV/atom, 原子间的力场收敛精度设置为0.1 eV/nm, 最大位移设置为5×10-5nm, 最大应力设置为0.02 GPa. 采用平面波与原子轨道线性组合法相结合, 计算体系中的密立根(Mulliken)电荷和键布居数[19, 20].
3 结果与讨论
3.1 表面原子几何结构
根据上面八种表面原子终止结构, 分别建立了32个原子的晶体表面原胞, 其化学式均为Ag12P4O16, 将它们置入一个方形空腔中, 选取真空厚度为0.6 nm, 初始晶胞参数a0=b0=0.8645 nm, r=120o, 这里分别采用LDA-CAPZ和GGA-PBE方法计算体系的总能量, 其结果如表1所示.
对于Ag3PO4(111)面的八种终止原子结构模型, 它们总的原子数相同, 同种原子个数也相同, 在仅考虑近邻成键情况下, 表面内层原子所处的环境结构相同, 但外表层原子的环境却有很大的 区别, 因而八种模型的体系总能量也应不同. 这八种结构模型的体系总能量中大部分计算结果较好地达到了设定的收敛度. 可以看出, 表面原子几何结构优化后, 用LDA-CAPZ和GGA-PBE方法计算的B类表面结构体系的总能量比其它七种表面结构体系的总能量分别低0.2436—15.3238 eV和0.2700—8.7706 eV. 根据总能量最小原理, B类表面原子终止结构是Ag3PO4(111)面最可能出现的表面原子几何构型, 如图2所示. 从而对文献[12]和[13]中关于该表面原子终止结构从能量上得到解释.

表1 八种表面原子终止结构模型的体系总能量和平均原子能量收敛度
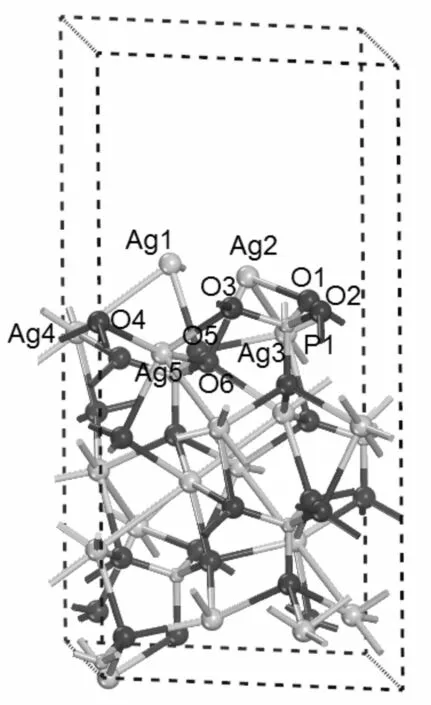
图2 立方相Ag3PO4(111)面24层模型图. 黑色球、小灰球、大灰球分别表示O、P和Ag原子Fig. 2 The 24-layer model of cubic Ag3PO4(111) surface. Black, small gray and big gray spheres represent the O, P and Ag atoms, respectively
3.2 表面能
在建立的表面模型中, 真空厚度和原子层数对表面能和表面弛豫的计算结果有很大关系[21,22]. 由于参与弛豫的原子层可能不仅仅限于表面几层, 为了获得可靠的结果, 重建了B类表面原子结构模型, 分别考虑不同原子层数和不同真空厚度的表面能收敛情况. 具体通过表面原子几何结构优化, 获得稳定的表面结构, 最终以确定合适的表面结构模型参数. 表面能定义为
Esurf=(Eslab-NEbulk)/2A,
(1)
式中N为表面单胞所含的Ag3PO4分子数; Esurf为含有N个分子的表面所具有的表面能; Eslab为含有N个Ag3PO4分子的表面单胞能量; Ebulk为体相中每个Ag3PO4分子的能量; 2A为表面单胞上下总表面积.
首先采用LDA-CAPZ和GGA-PBE计算方法研究了原子层数为16的B类表面模型的表面能, 发现真空厚度为0.4 nm时, 这两种方法计算出的表面能收敛度均达到了0.01 J/m2, 这与文献[21]中在研究金红石型TiO2(110)面时情况一致.
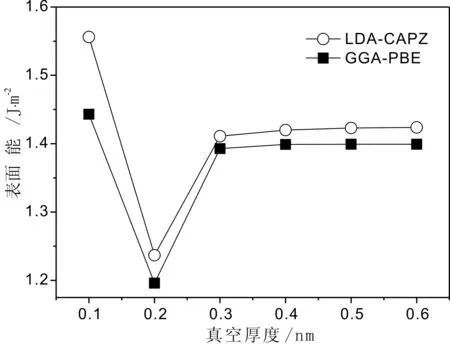
图3 原子层数为16层时表面能与真空厚度的关系Fig. 3 Dependence of surface energies on vacuum space thickness with atomic layer of 16
图3显示了优化后的表面能与真空厚度的关系. 从图3中看到当真空厚度小于0.3 nm时表面能的变化很大, 说明了当真空厚度小于或者接近于晶体内原子间的平衡距离时(0.2 nm左右), 模型内真空分隔的上下表面之间表现出很强相互作用. 为了减少真空厚度在研究表面层数与表面能关系的影响, 我们选取了更厚的真空层(0.6 nm)来消除两表面间的相互作用.
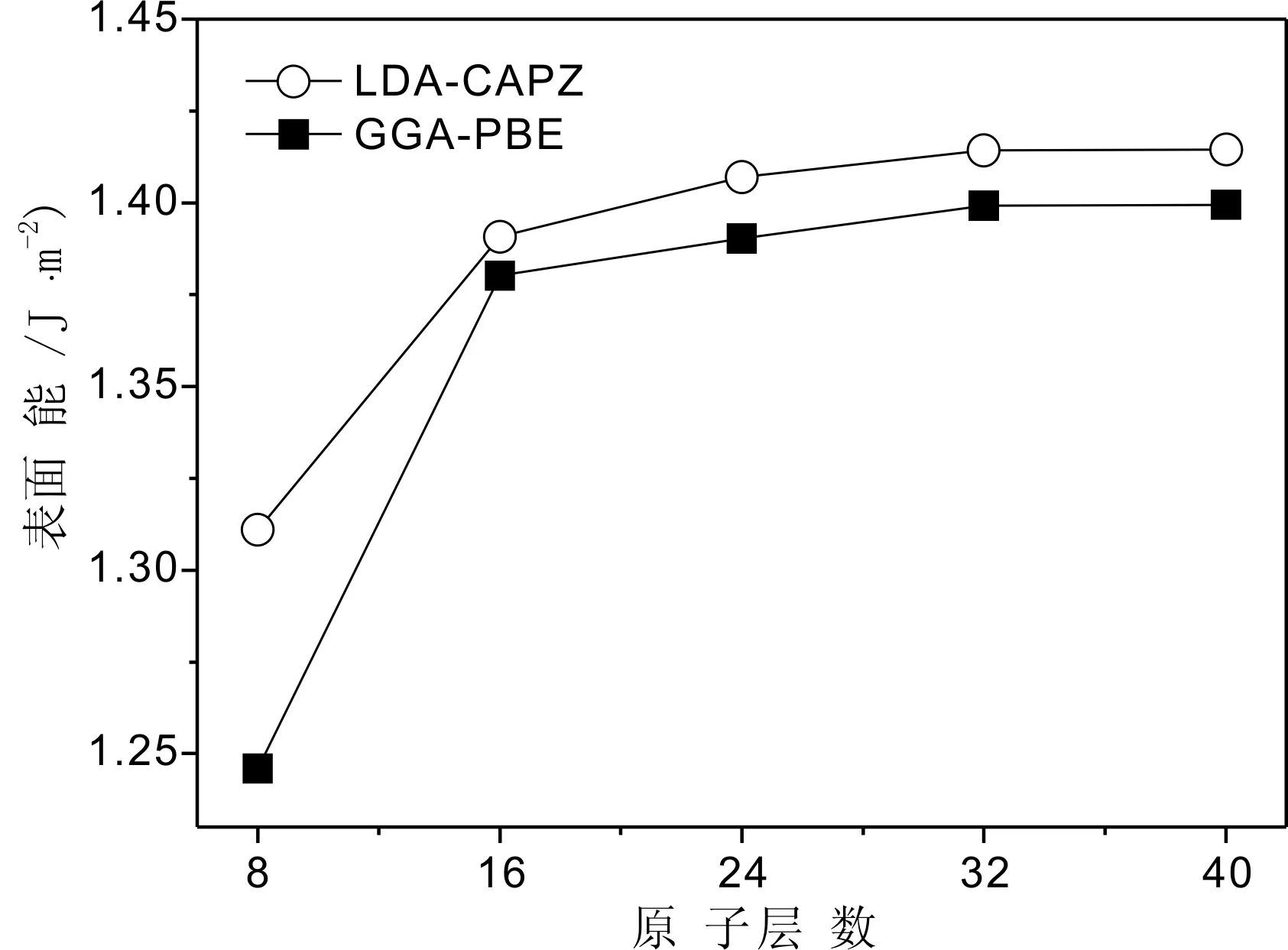
图4 真空层厚度为0.6 nm时表面能与原子层数的关系Fig. 4 Dependence of surface energies on the number of atomic layer with vacuum space thickness of 0.6 nm
从图4中可以看到, 表面能与建立的表面层数有较大的关系. 当原子层数达到24层时, 表面能收敛于1.41 J/m2(LDA-CAPZ)和1.39 J/m2(GGA-PBE), 收敛度小于0.002 J/m2, 这个结果稍小于文献[12]和[13]中用其他方法计算的表面能为1.65 J/m2. 将他们的模型与我们前面的八种表面结构模型相比较, 很容易发现他们认定的Ag3PO4(111)表面原子几何结构实际为本文中A类结构模型. 事实证明, B类结构模型比A类结构模型具有更低的表面能, 即具有更稳定的结构. 我们计算的A类结构的表面能约为1.53 J/m2, 比B类结构的表面能稍大. 这里没有发现像金红石型TiO2(110)面出现的表面能与原子层数关系那样的奇偶行为[21]. 而对于原子层数为16的B类表面模型, 当真空厚度为0.6 nm时,其表面能为1.94 J/m2.对该表面原子几何结构优化后, 表面原子发生了明显的弛豫现象, 表面能有较大幅度的降低, 表面结构趋于更稳定.
3.3 表面原子弛豫


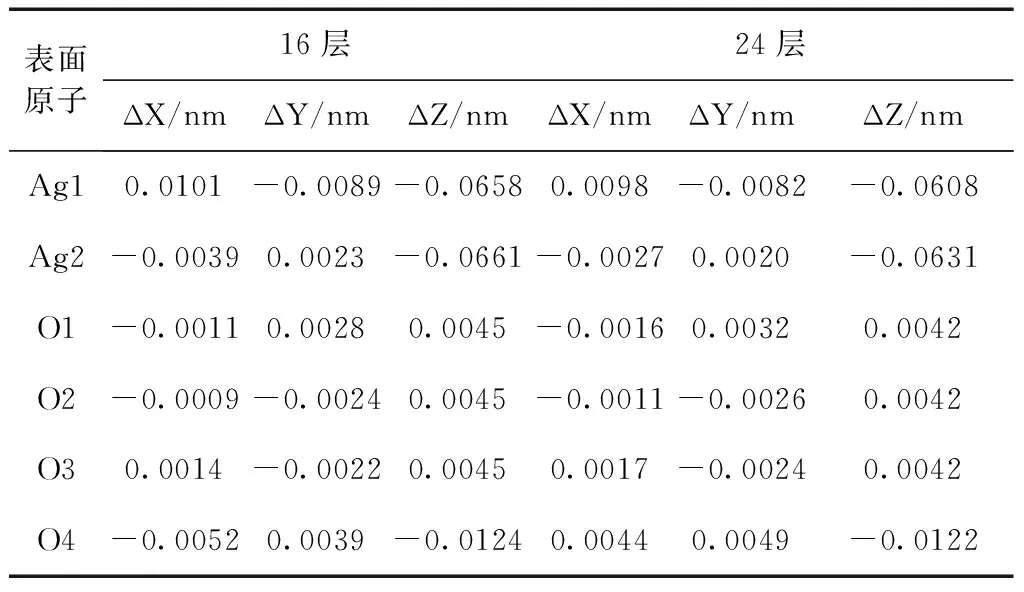
表面原子16层24层ΔX/nmΔY/nmΔZ/nmΔX/nmΔY/nmΔZ/nmAg10.0101-0.0089-0.06580.0098-0.0082-0.0608Ag2-0.00390.0023-0.0661-0.00270.0020-0.0631O1-0.00110.00280.0045-0.00160.00320.0042O2-0.0009-0.00240.0045-0.0011-0.00260.0042O30.0014-0.00220.00450.0017-0.00240.0042O4-0.00520.0039-0.01240.00440.0049-0.0122

表3列出了理想表面和弛豫表面原子间的键长及键布居数. 从表中可以看出, 弛豫后表面几层原子的键长发生了较大的变化, 键长均变短. 其中Ag1—O5键长变化最为明显, 收缩了0.0263 nm, 同时O4—Ag5键长也有较大的收缩, 使其成为表面O—Ag键中最短的键长, 仅有0.2129 nm, 这可能与O4失去上方的一个配位有关. 由于半径较大的Ag离子电子云易被周围极化力较强的O离子极化, 表面Ag离子失去上方键连的O离子, 其对称性降低, 离子极化程度比体相中相对要高. 根据键型变异原理, 离子的极化程度越高, 其键能越大, 键长应该越短, 这可能是导致与Ag离子相连的Ag1—O5、Ag2—O1和Ag2—O6键在弛豫后, 出现键长缩短的主要原因. 由于O离子半径比Ag离子半径大, 而且弛豫后表面O离子中心位于Ag3PO4(111)面的最表面处. 因而暴露在Ag3PO4(111)面的最表面是未满配位的O原子.
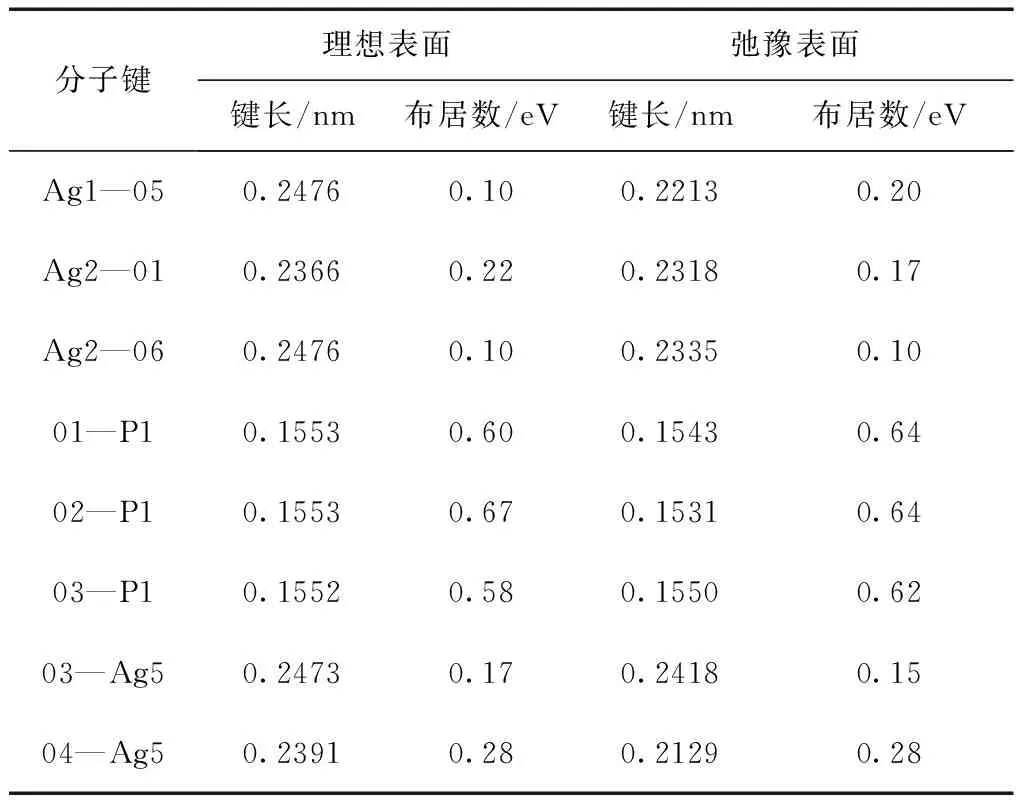
表3 原子间键长及键布居数
立方相Ag3PO4(111)面的表面原子发生弛豫后, 其表面原子上的核外电荷分布将发生改变. 表4为理想表面和弛豫表面通过Mulliken布居分析后的表面各原子核外电荷电量. 可以看出, 弛豫后Ag1和Ag2原子的核外电荷电量分别增加了0.01 和0.02, 而O4原子的核外电荷电量也增加了0.09, 这与他们向表面内部弛豫有关. 而O1、O2和O3原子的核外电荷电量分别减少了0.02—0.03, 这些减少的负电荷转移到了 P1原子上, 使P1原子的核外电荷电量增加了约0.06, 而其它原子没有发生明显的净电荷转移. 总体上表现出表面悬挂键上多余的电荷向表面内层原子转移, 导致表面原子之间的相互作用也随之发生改变, 体系总能量降低.

表4 Mulliken净电荷布居数 (单位电量)
4 结 论
采用密度泛函理论的平面波超软赝势方法获得Ag3PO4(111)面原子几何和原子弛豫结构的数据.通过对八种不同原子终止结构的体系总能量进行计算, 发现B类表面结构最为稳定. 随后进一步对该结构进行原子几何结构弛豫, 结果显示了表面的Ag原子向表面里移动, 而次层的O1、O2和O3原子则向表面外移动, 使原来位于Ag原子下方的O原子移动到最表面. 因此, Ag3PO4(111)面应终止于三个O原子, 其中两个为三配位, 一个为二配位, 次层为两个Ag原子, 均为三配位. 而且表面原子的弛豫,使得表面原子的核外电荷发生了重新分布, 即表面悬挂键上多余的电荷向表面内层转移. 总体来说, 表面原子几何结构弛豫与建立的表面模型有较大关系, 并通过结构优化以及体系能量的比较, 证实了这里获得的表面原子弛豫结果是可信的.
[1] Kudo A, Mesiki Y. Heterogeneous photocatalyst materials for water splitting [J].Chem.Soc.Rev., 2009, 38: 253.
[2] Inoue Y. Photocatalytic water splitting by RuO2-loaded metal oxides and nitrides with d0- and d10-related electronic configurations [J].EnergyEnviron.Sci., 2009, 2: 364.
[3] Lang X J, Chen X D, Zhao J C. Heterogeneous visible light photocatalysis for selective organic transformations [J].Chem.Soc.Rev., 2014, 43: 473.
[4] Pan C S, Li D, Ma X G,etal. Effects of distortion of PO4tetrahedron on the photocatalytic performances of BiPO4[J].Catal.Sci.Technol., 2011, 1: 1399.
[5] Lu B, Ma X G, Pan C S,etal. Photocatalytic and photoelectron- chemical properties of in situ carbon hybridized BiPO4films [J].AppliedCatalysisA,General, 2012, 93: 435.
[6] Yi Z G, Ye J H, Kikugawa N,etal. An orthophosphate semiconductor with photooxidation properties under visible-light irradiation [J].Nat.Mater., 2010, 9: 559.
[7] Bi Y P, Hu H Y, Ouyang S X,etal. Selective growth of Ag3PO4submicro-cubes on Ag nanowires to fabricate necklace-like heterostructures for photocatalytic applications [J].J.Mater.Chem., 2012, 22: 14847.
[8] Bi Y P, Ouyang S X, Umezawa N,etal. Facet effect of single-crystalline Ag3PO4sub-microcrystals on photocatalytic properties [J].J.Am.Chem.Soc., 2011, 133: 6490.
[9] Umezawa N, Ouyang S X, Ye J H. Theoretical study of high photocatalytic performance of Ag3PO4[J].Phys.Rev. B, 2011, 83: 035202.
[10] Ma X G, Lu B, Li D,etal. Origin of photocatalytic activation of silver orthophosphate from first-principles [J].J.Phys.Chem. C, 2011, 115(11): 4680.
[11] Yang H G, Sun C H, Qiao S Z,etal. Anatase TiO2single crystals with a large percentage of reactive facets [J].Nature, 2008, 453: 638.
[12] Martin D J, Umezawa N, Chen X W,etal. Facet engineered Ag3PO4for efficient water photooxidation [J].EnergyEnviron.Sci., 2013, 6: 3380.
[13] Zheng B J, Wang X, Liu C,etal. High-efficiently visible light-responsive photocatalysts: Ag3PO4tetrahedral microcrystals with exposed {111} facets of high surface energy [J].J.Mater.Chem. A, 2013, 1: 12635.
[14] Ng H N, Calvo C, Faggiani R. A new investigation of the structure of silver orthophosphate [J].ActaCrystallogr. B, 1978, 34: 898.
[15] Vanderbilt D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism [J].Phys.Rev. B, 1990, 41: 7892.
[16] Ceperley D M, Alder B J. Ground state of the electron gas by a stochastic method [J].Phys.Rev.Lett., 1980, 45: 566.
[17] Perdew J P, Burke K, Ernzerhof M. Generalized gradient approximation made simple [J].Phys.Rev.Lett., 1996, 77: 3865.
[18] Monkhorst H J, Pack J D. Special points for brillouin-zone integrations [J].Phys.Rev. B, 1976, 13: 5188.
[19] Sanchez-Portal D, Artacho E, Soler J M. Projection of plane-wave calculations into atomic orbitals [J].SolidStateCommun., 1995, 95: 685.
[20] Segall M D, Shah R, Pickard C J,etal. Population analysis of plane-wave electronic structure calculations of bulk materials [J].Phys.Rev. B, 1996, 54: 16317.
[21] Bate S P, Kresse G, Gillan M J. A systematic study of the surface energetics and structure of TiO2(110) by first-principles calculations [J].Surf.Sci., 1997, 385: 386.
[22] Diebold U, Ruzycki N, Herman G S,etal.One step towards bridging the materials gap: surface studies of TiO2anatase [J].CatalysisToday, 2003, 85: 93.
First-principles calculations on the geometry and relaxation structure of cubic Ag3PO4(111) surface
MA Xin-Guo1,2, LIU Na1, ZHU Lin1, XU Guo-Wang1,2, HUANG Chu-Yun1,2
(1. School of Science, Hubei University of Technology, Wuhan 430068, China; 2. Hubei Collaborative Innovation Center for High-efficiency Utilization of Solar Energy, Hubei University of Technology, Wuhan 430068, China)
First-principles calculations based on the plane-wave ultrasoft pseudopotential method have been taken to investigate the surface energy and structure of cubic Ag3PO4(111) surface. There are eight different structures of cubic Ag3PO4(111) surface because different atoms are terminated on the surface layers. The calculated results show that the B model is much more stable than other seven structures. By investigating the effect of variable vacuum width and slab thickness on the surface energies and surface atomic displacements, the surface energies are converged to 1.41 J/m2(LDA-CAPZ) and 1.39 J/m2(GGA-PBE) for slab thickness of 24-atom layers and vacuum width of 0.6 nm. At last, by the atomic relaxation of surface structure, the two threefold coordinated Ag atoms are inward relaxation of more 0.06 nm, and three O atoms in sublayer are outward relaxation of about 0.0042 nm. So O atoms are exposed to the outermost surface, while part of electrons upon O atoms transfer from the surface to internal atoms, the structure becomes more stable.This study serves as an initial and important step toward more in-depth analysis of the photocatalytic activity origin of Ag3PO4surface.
First-principles; Silver orthophosphate; Surface structure; Relaxation
103969/j.issn.1000-0364.2015.12.026
2014-12-11
国家自然科学基金(51102150,51472081);中国博士后基金(201104085);湖北工业大学高层次人才启动基金(GCRC13014)
马新国(1978—),男,湖北汉川人,博士,副教授. E-mail: maxg2013@sohu.com
黄楚云. E-mail: chuyunh@163.com
O485
A
1000-0364(2015)06-1064-07
猜你喜欢
东北水利水电(2022年6期)2022-06-28
科学技术创新(2022年15期)2022-05-18
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
青岛大学学报(工程技术版)(2019年2期)2019-09-10
电子制作(2019年11期)2019-07-04
当代陕西(2019年6期)2019-04-17
枣庄学院学报(2015年5期)2016-01-09
中学化学(2015年8期)2015-12-29
小天使·五年级语数英综合(2015年4期)2015-04-20
外语学刊(2014年3期)2014-12-03
