
钛乙烯储氢材料的热力学和动力学性质研究
2015-03-22 10:42何娜,高涛
原子与分子物理学报 2015年6期
何 娜, 高 涛
(1.咸宁职业技术学院工学院, 咸宁 437000; 2.四川大学原子与分子物理研究所, 成都 610065)
钛乙烯储氢材料的热力学和动力学性质研究
何 娜1, 高 涛2
(1.咸宁职业技术学院工学院, 咸宁 437000; 2.四川大学原子与分子物理研究所, 成都 610065)
之前的工作中已经预测了钛乙烯(C2H4Ti)储氢的几何结构,本文将继续利用密度泛函理论(DFT)和B3LYP杂化密度泛函方法来计算之前预测出的C2H4Ti(H2)n结构中的C2H4Ti和H2反应的焓变和自由能变.通过焓变和自由能变的计算结果,可以看出:钛乙烯在298 K、250 K、200 K下可以稳定的吸附5个氢分子,同时放出大量的热量,生成的C2H4Ti(H2)n化合物在常温下也具有热力学稳定性.此外,本文通过Gaussian03中的伯恩近似分子动力学(BOMD)方法计算了C2H4Ti(H2)5化合物在298 K、250 K、200 K三个温度下的动力学性质.通过动力学研究的结果,可以发现钛乙烯分子在常温298 K下储氢时间不长;同时得到钛乙烯分子在200 K下能长时间稳定吸附五个氢分子,这进一步表明降低温度对钛乙烯分子稳定储氢是有利的.
钛乙烯; 焓变; 自由能变; 伯恩近似分子动力学
1 引 言
当今,氢能作为一种丰富的、可更新的、清洁安全的、很经济的新型能源,被广泛的视为将来可替代石油燃料的最佳候选者之一.而钛乙烯(C2H4Ti)作为一种储氢量很高的新型储氢材料受到了广泛关注.近几年来,Durgun[1]等人通过第一性原理的计算用周期性边界条件预言了C2H4Ti2体系可以吸附10个氢分子,并且得到Ti在过渡金属中和C2H4组成的化合物的储氢量是相当高的,同时结合能也很合理.除此之外,实验上A.B.Phillips[2]等人利用基于高分辨率重力技术的表面声波共振器测得C2H4Ti的储氢动力学曲线和储氢量,实验结果表明C2H4Ti储氢量可高达约12wt%,这些与理论预测的相一致.这说明C2H4Ti作为一种新型的储氢材料,确实具有最优性能.
在前期的工作中,我们采用了与前人不同的方法:密度泛函理论(DFT)和交换泛函方法(B3LYP),通过结构优化得到了C2H4Ti储氢的两个同分异构体,并通过动力学稳定性的分析得到了最稳定的C2H4Ti储氢构型[3].现在,我们将继续前期的工作,在最稳定的C2H4Ti储氢构型的基础上,通过Gaussian03程序中热力学的基本公式,得到了C2H4Ti储氢反应的标准摩尔反应焓变、标准吉布斯反应自由能变、标准摩尔生成焓变和标准吉布斯生成自由能变这些热力学数据.我们通过对这些热力学数据的详尽分析来推断C2H4Ti分子在298 K,一个标准大气压下能否稳定的储氢.此外,我们还通过伯恩近似分子动力学(BOMD)方法得到了C2H4Ti(H2)5化合物分别在298 K、250 K、200 K温度下的动力学曲线图.通过对这些动力学曲线图的分析,我们可以得到C2H4Ti分子能稳定吸附5个氢分子的时间,从而推断出温度的高低对C2H4Ti分子储氢时间的影响.
2 计算方法
在高斯中用来计算热化学数据的等式和热力学的标准书中给出的公式是等价的(这在McQuarrie和Simon的分子热力学书中讨论过).因此,在本篇文章中,我们选用高斯中的热力学方法[4-6]计算了C2H4Ti和H2反应的标准摩尔焓变ΔrH°(298 K)和标准吉布斯自由能变ΔrG°(298 K),计算公式如下:
ΔrH°(298 K)=∑(ε0+Hcorr)C2H4Ti(H2)n
-∑(ε0+Hcorr)C2H4Ti-n∑(ε0+Hcorr)H2
(1)
ΔrG°(298 K)=∑(ε0+Gcorr)C2H4Ti(H2)n
-∑(ε0+Gcorr)C2H4Ti-n∑(ε0+Gcorr)H2
(2)
其中ε0表示电子总能量,Hcoor表示温度焓的值,Gcoor表示温度自由能的值.
同时,我们选用高斯中的热力学方法计算了C2H4Ti(H2)n的标准摩尔生成焓变和标准吉布斯生成自由能变.一般计算一种未知物质的生成焓有两种方法,第一种方法是原子反应方法[7],可以分成两步进行:第一步,计算物质在0 K反应时的生成焓ΔfH°(0 K),第二步,计算物质在298 K时的生成焓ΔfH°(298 K).第二种方法是键分裂独立反应,这种方法适用于那些已经知道生成热实验值的物质.因为我们计算的C2H4Ti(H2)n的实验数据并不充分,因此我们选用了第一种方法来进行计算.
根据高斯中热力学计算公式[8,9],我们可以得到C2H4Ti(H2)n的标准摩尔生成焓变和标准吉布斯生成自由能变的公式为:
(3)
ΔfH°(M,298 K)=ΔfH°(M,0 K)+(H°M(298 K)
(4)
ΔfG°(M,298 K)=ΔfH°(298 K)-
298.15(S°(M,298 K)-∑S°(X,298 K))
(5)
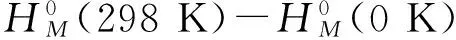
此外,为了进一步研究C2H4Ti分子在常温下储氢的稳定性,我们通过Gaussian03中的伯恩近似分子动力学(BOMD)方法[11-16]来计算C2H4Ti(H2)5化合物在298 K﹑250 K﹑200 K三个温度下的动力学性质.
3 结果与讨论
3.1 C2H4Ti储氢材料的热力学性质
近几年来,越来越多的人运用量子化学来研究物质的热力学性质[17-19],在本节中我们选用高斯中的热力学方法计算了C2H4Ti和H2反应的标准摩尔焓变和标准吉布斯自由能变,以及标准摩尔生成焓变△fH0和标准吉布斯生成自由能变△fG0.
根据高斯中热力学计算公式(1)、(2),我们可求得在298 K、1标准大气压条件下,C2H4Ti和H2反应的焓变和吉普斯自由能变的数据如下表1所示:
表1 C2H4Ti和5个H2反应的标准摩尔焓变和标准吉普斯自由能变
Table 1 Calculated enthalpies and Gibbs free energies of reaction for the reactions C2H4Ti+nH2→C2H4Ti(H2)nat 298 K

ReactionΔrH°(298K)/kcal/molΔrG°(298K)/kcal/molC2H4Ti+H2-37.65-31.38C2H4Ti+2H2-43.93-25.10C2H4Ti+3H2-43.93-18.83C2H4Ti+4H2-43.93-12.55C2H4Ti+5H2-25.1012.55
通过表1中热力学数据,可以看出C2H4Ti和这5个H2的标准摩尔反应焓变ΔrH°(298 K)均为负数,表明C2H4Ti+nH2→C2H4Ti(H2)n反应是沿着正方向进行的,并且该反应为放热反应,因此降低温度对该反应平衡有利.同时C2H4Ti和前4个H2反应的标准吉布斯自由能变ΔrG°(298 K)均为负值,表明在298 K下C2H4Ti+nH2→C2H4Ti(H2)n的反应可以自发进行.而C2H4Ti和第5个H2反应的标准吉布斯自由能变为正值,这是因为第五个H2在C2H4Ti上的吸附很弱,C2H4Ti+5H2→C2H4Ti(H2)5的反应在298 K下不易自发进行.但又因为此反应的标准吉布斯自由能正值很小,同时标准吉布斯自由能变ΔrG°(298 K)不仅和反应温度有关,也和反应压强有关,所以稍微调节下温度和分压,则可使ΔrG°(298 K)<0,该反应即可正向自发进行.
为了进一步测试C2H4Ti+5H2→C2H4Ti(H2)5反应与温度的关系,我们将温度调节到250 K和200 K时,得到了C2H4Ti和5个H2反应的标准 摩尔焓变和标准吉普斯自由能变的数据如下表2.可以看出在250 K和200 K温度下,C2H4Ti+5H2→C2H4Ti(H2)5反应的标准摩尔焓变ΔrH°(298 K)和标准吉布斯自由能变ΔrG°(298 K)均为负值,因此C2H4Ti和5个H2的放热反应在250K和
表2 C2H4Ti+5H2→C2H4Ti(H2)5在250 K、200 K下的标准反应焓变和自由能变
Table 2 Calculated enthalpies and Gibbs free energies of reaction C2H4Ti+5 H2→C2H4Ti(H2)5at 250 K and 200 K

反应(C2H4Ti+5H2)温度ΔrH°/(kcal/mol)ΔrG°/(kcal/mol)250K-37.65-1.26200K-31.38-6.28
200 K温度下可以自发的进行.
根据高斯中热力学计算公式(3)—(5),我们可求得C2H4Ti(H2)n的标准摩尔生成焓变和标准吉布斯生成自由能变.同时根据计算化学对比与基准数据库[10]中给出的C、H、Ti各元素的生成焓和原子的熵的实验值如下表:
表3 C、H、Ti各元素的生成焓和原子的熵的实验值
Table 3 Experimental enthalpies of formation of elements and entropies of atoms
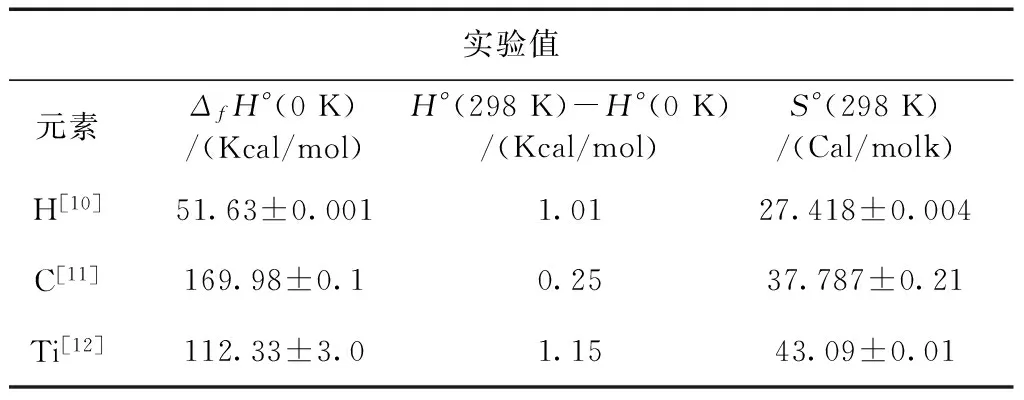
实验值元素ΔfH°(0K)/(Kcal/mol)H°(298K)-H°(0K)/(Kcal/mol)S°(298K)/(Cal/molk)H[10]51.63±0.0011.0127.418±0.004C[11]169.98±0.10.2537.787±0.21Ti[12]112.33±3.01.1543.09±0.01
因此我们通过C、H、Ti各元素在0 K下的生成焓和原子的熵的实验值,可求得在298 K、1标准大气压条件下,C2H4Ti(H2)n各物质的标准摩尔生成焓变和标准吉普斯生成自由能变的数据如下表4所示:
表4 C2H4Ti(H2)n在298 K下的标准摩尔生成焓变和吉普斯生成自由能变
Table 4 Calculated enthalpies and Gibbs free energy of formation of C2H4Ti(H2)nat 298 K
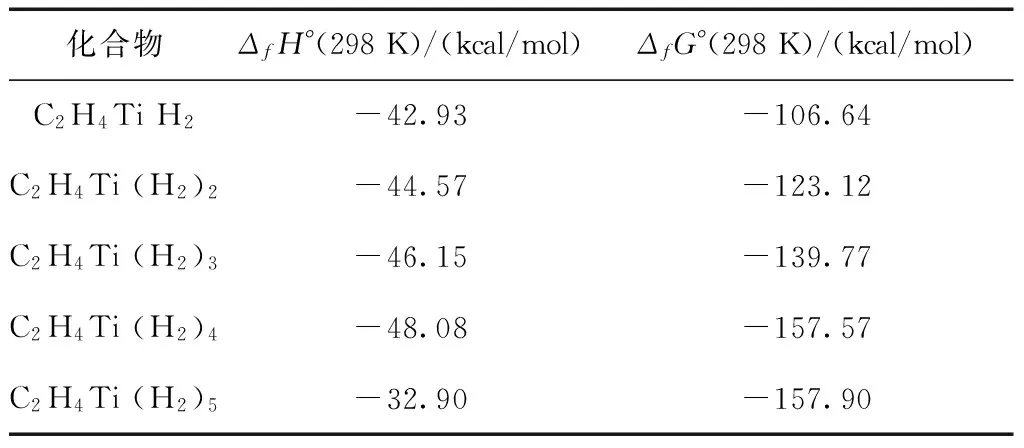
化合物ΔfH°(298K)/(kcal/mol)ΔfG°(298K)/(kcal/mol)C2H4TiH2-42.93-106.64C2H4Ti(H2)2-44.57-123.12C2H4Ti(H2)3-46.15-139.77C2H4Ti(H2)4-48.08-157.57C2H4Ti(H2)5-32.90-157.90
通过表4,我们可以看到标准摩尔生成焓变ΔfH°(298 K)和标准吉布斯生成自由能变ΔfG°(298 K)都为负值,表明C2H4Ti(H2)n化合物在298 K、1个标准大气压条件下是可以稳定生成的.同时我们发现随着C2H4Ti分子吸附氢分子数量的增多,标准吉普斯生成自由能变ΔfG°(298 K)的值是逐渐减小的,因为反应总是朝着自由能减小的方向进行,所以C2H4Ti+nH2→C2H4Ti(H2)n在常温下是可以自发进行的.
3.2 C2H4Ti储氢材料的动力学性质
为了进一步研究C2H4Ti分子在常温下储氢的稳定性,我们通过Gaussian03中的伯恩近似分子动力学(BOMD)方法[11-16]来计算C2H4Ti(H2)5化合物在298 K﹑250 K﹑200 K三个温度下的动力学性质.为了使氢分子随时间变化的轨迹更清晰,我们指定每一个轨迹中的最大步骤数为3000步,设定动力学计算的步长为1飞秒,设置混合泛函因子的参数为IOP(1/44=518573).同时我们对每个路径指定了六个终止标准:1. 每对片段质心间的最小距离至少大于3.0 Bohr;2.不同片段中的任何原子间的最小距离大于1 Bohr;3.任一原子与所在片段质心的最大距离至少小于800 Bohr;4.同一片段内任意两原子间最大距离至少小于800 Bohr;5.片段的梯度小于(10-6) Hartrees/Bohr;6.如果ITest=1,原子7Atom和8Atom的距离大于2.0 Bohr.在分析C2H4Ti(H2)5化合物的动力学性质时,我们可以发现随着时间的延长,最后C2H4Ti分子周围的4个氢分子中总有一个氢分子会被Ti原子吸附生成氢化物的形式.因此为了计算能顺利进行,我们将这个氢分子和C2H4Ti分子看成一个整体.因此我们在图中只给出了未和C2H4Ti分子形成化学键的其他四个氢分子的运动轨迹.
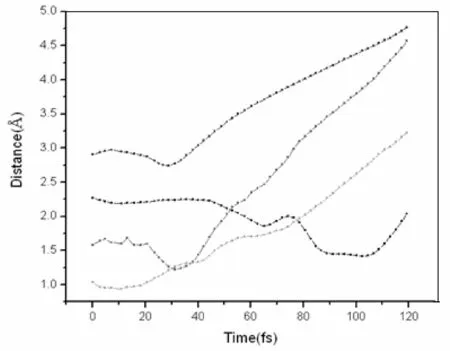
图1 C2H4Ti(H2)5化合物在298 K温度下的动力学轨迹图Fig. 1 Trajectories of time evolution of RH2 in C2H4Ti-5H2 complex at 298 K
由图1我们可以看出未成键的四个氢分子在前20 fs内和体系的距离基本上保持不变,它们是可以稳定吸附在体系周围的.但是我们可以看到当时间超过20 fs且逐渐增大时,这四个氢分子与体系的距离开始有个先减小后增大的过程,这表明氢分子在体系周围振动,而随着时间的进一步延长,我们会发现这四个氢分子总的趋势是与体系的距离越来越大,这说明这四个氢分子逐渐脱离了体系,被释放了出来.其中我们还可以看到蓝线表示的Ti原子顶上的氢分子与体系距离增大的最快,这说明Ti原子顶上的氢分子最不稳定,这是因为我们前面结构分析中看到的:Ti原子与顶上的氢分子间距最大,它们的键能也很小,C2H4Ti分子对其吸附本来就很弱.所以我们可以得到C2H4Ti分子在298 K下能稳定吸附一个氢分子,其他四个氢分子都不能长时间被稳定吸附,且Ti原子顶上的氢分子最先被释放出来.这与Yasuharu Okamoto用ADMP-MD方法计算得到的结果一致[20].
接下来我们通过BOMD得到了C2H4Ti(H2)5化合物在250 K温度下的动力学轨迹.
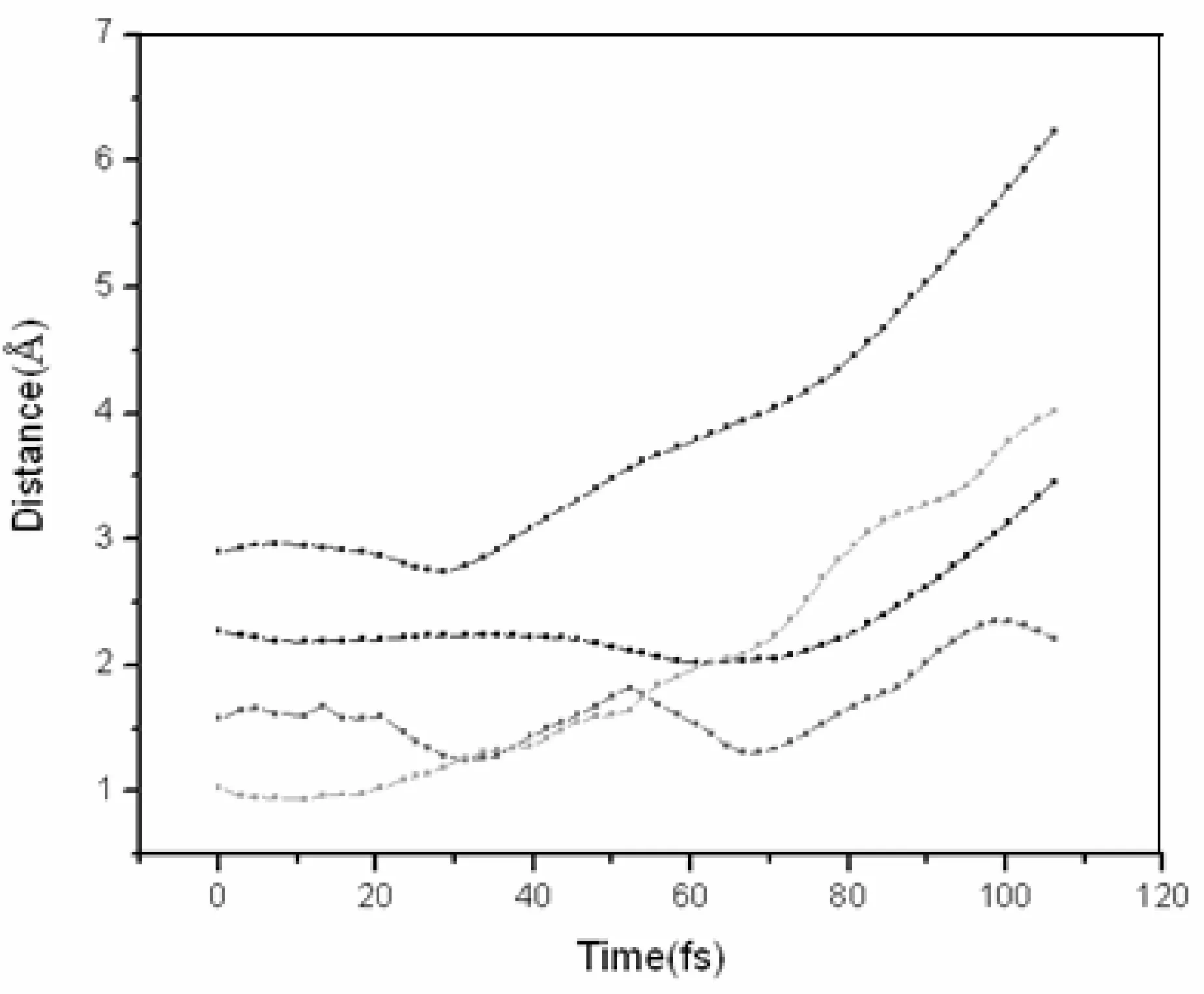
图2 C2H4Ti(H2)5化合物在250 K温度下的动力学轨迹图Fig. 2 Trajectories of time evolution of RH2 in C2H4Ti-5H2 complex at 250 K
由图2我们可以看出未成键的四个氢分子中的红﹑绿﹑黑三个氢分子和体系的距离的变化是很缓慢的,在60 fs之前我们可以认为还是被吸附在体系周围的.同时红线所表示的氢分子一直是在体系周围振动的,因此我们认为它是可以稳定的吸附在体系周围的.而蓝线所表示的Ti原子顶上的氢分子,它与体系的距离还是随着时间的增加逐渐增大的,因此它还是不能被稳定吸附在体系的周围.所以我们可以得到C2H4Ti分子在250 K下能稳定吸附两个氢分子,其他三个氢分子虽然都不能被稳定吸附,但要比298 K下稳定的时间长些,这也表明降低温度对C2H4Ti分子稳定储氢是有利的,且Ti原子顶上的氢分子还是最先被释放出来.
最后我们通过BOMD得到了C2H4Ti(H2)5化合物在200 K温度下的动力学轨迹.
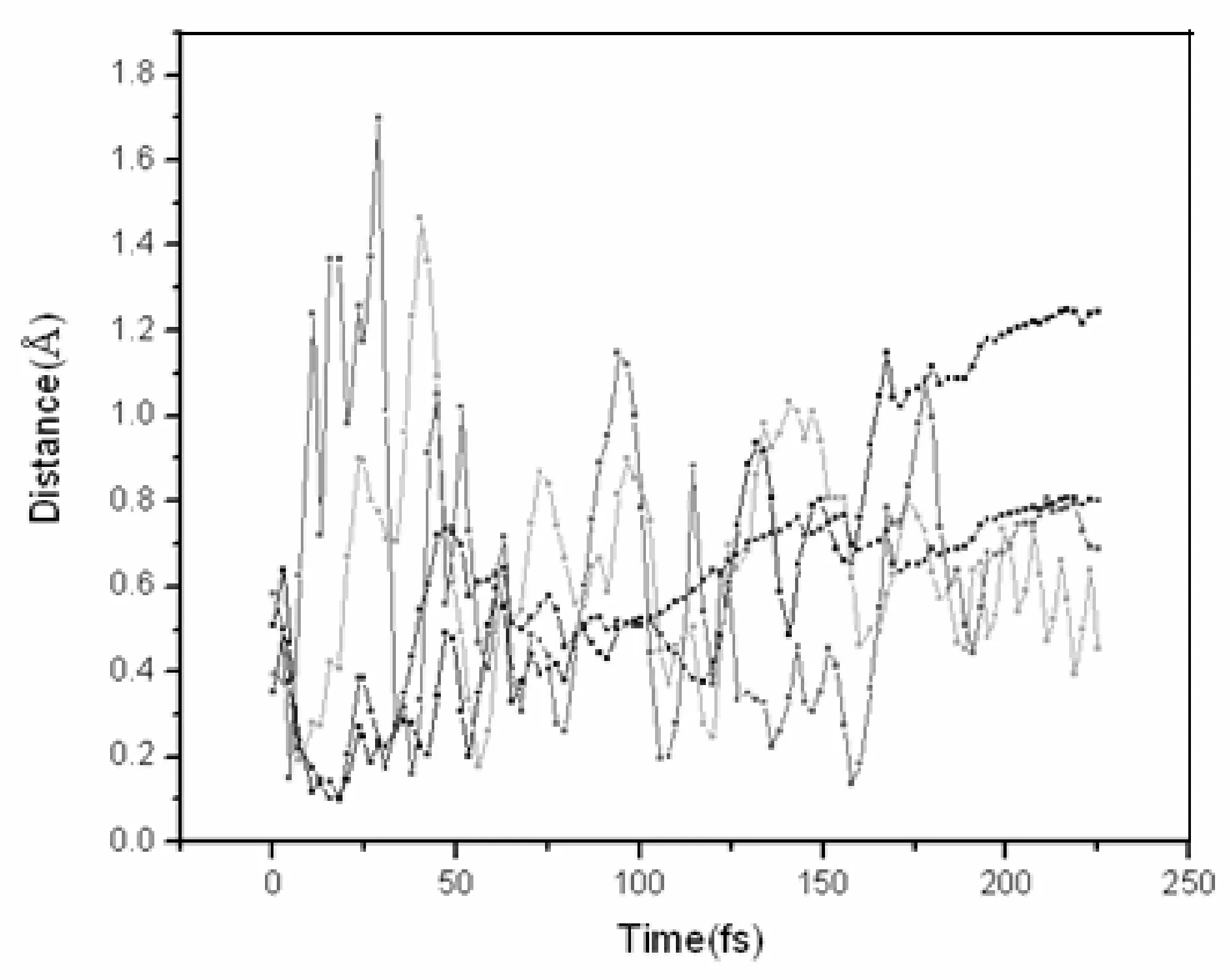
图3 C2H4Ti(H2)5化合物在200 K温度下的动力学轨迹图Fig. 3 Trajectories of time evolution of RH2 in C2H4Ti-5H2 complex at 200 K
由图3我们可以看出未成键的四个氢分子与体系的距离随着时间的增加是在上下波动的.这表明这四个氢分子在体系周围振动,并不会远离体系而去,因此我们认为这四个氢分子是可以稳定的吸附在体系周围的.这时蓝线所表示的Ti原子顶上的氢分子虽然波动相对较小,但其并没有像298 K﹑250 K时的那样,而是随着时间的增加其与体系的距离并未一直增大,因此我们认为顶上的氢分子此时也是稳定吸附的.所以我们可以得到C2H4Ti分子在200 K下能长时间稳定吸附五个氢分子,这进一步表明降低温度对C2H4Ti分子稳定储氢是有利的,这也就是我们在热力学部分所说的该反应为放热反应,则降低温度对该反应平衡有利.
4 结 论
本文采用Gaussian03程序中热力学的基本计算公式,以及计算化学对比与基准数据库(CCCBDB)中的实验数据,我们第一次给出了C2H4Ti储氢的标准摩尔反应焓变﹑标准吉布斯反应自由能变﹑标准摩尔生成焓变﹑标准吉布斯生成自由能变这些热力学数据.通过对这些热力学数据的分析表明C2H4Ti储氢反应是个放热反应,随着温度的降低C2H4Ti储氢反应更易进行,也就是说温度越低C2H4Ti储氢更稳定.
同时采用Gaussian03程序中伯恩近似分子动力学(BOMD)方法,我们第一次给出了C2H4Ti(H2)5在298 K﹑250 K﹑200 K温度下储氢的动力学数据.通过这些动力学数据分析表明C2H4Ti在298 K和250 K下只能短时间稳定储氢,随着时间延长,最多只能储两个氢.而在200 K的低温下,C2H4Ti可以长时间稳定储氢.这进一步表明C2H4Ti储氢在低温下更稳定.
[1] Zhou W, Yildirim T, Durgun E,etal. Hydrogen absorption properties of metal-ethylene complex[J].Phys.Rev. B, 2007, 76: 085434.
[2] Philips A B, Shivaram B S. High capacity hydrogen absorption in transition metal-ethylene complexes observed via nanogravimetry[J].Phys.Rev.Lett., 2008, 100: 105505.
[3] He N, Gao T, Zhang Z H,etal. Theoretical study of structure stability vibrational frequencies and electronic properties of hydrogen storage in titanium-ethylene complex[J].JournalofMolecularStructure-Theochem., 2009, 916: 147.
[4] McQuarrie D A, Simon J D.Molecularthermodynamics[M]. America: University Science Books Press, 1999.
[5] Curtiss,etal. Gaussian-4 theory with reduced perturbation orders[J].J.Chem.Phys., 1997, 106:1063.
[6] Lamba J, Burgin T. Calculated enthalpy values[J].J.Am.Chem.Soc., 1995, 117: 11299.
[7] Joseph W O.ThermochemistryinGaussian[M]. Pittsburgh: Gaussian Inc., 2000.
[8] Meier R, Wellnitz D, Kim S J,etal. The NH and CH bands of comet C/1996 B2 (hyakutake) [J].Icarus., 1998, 136: 268.
[9] Chase M W, Davies C A, Downey J R,etal. JANAF thermochemical tables[J].J.Phys.Ref., 1985, 14: 1.
[10] http://cccbdb.nist.gov/default.htm
[11] Helgaker T, Uggerud E, Jensen H J A. Integration of the classical equations of motion on ab initio molecular potential energy surfaces using gradients and Hessians: application to translational energy release upon fragmentation[J].Chem.Phys.Lett., 1990, 173: 145.
[12] Uggerud E, Helgaker T. Dynamics of the reaction CH2OH+→CHO++H2. Translational energy release from ab initio trajectory calculations[J].J.Am.Chem.Soc., 1992, 114: 4265.
[13] Chen W, Hase W L, Schlegel H B. Ab initio classical trajectory study of H2CO→H2+CO dissociation[J].Chem.Phys.Lett., 1994, 228: 436.
[14] Millam J M , Bakken V, Chen W,etal. Theoretical methods and algorithms-ab initio classical trajectories on the Born-Oppenheimer surface: Hessian-based integrators using fifth-order polynomial and rational function fits[J].J.Chem.Phys., 1999, 111: 3800.
[15] Li X, Millam J M, Schlegel H B.Gaussian03,RevisionC.02[M]. Wallingford CT: Gaussian Inc., 2004.
[16] Hase W L, Duchovic R J, Hu X,etal. Program exchange[J].J.QuantumChem., 1996, 16: 671.
[17] Denis P A, Ventura O N. Density functional investigation of atmospheric sulfur chemistry. I. Enthalpy of formation of HSO and related molecules[J].Int.J.QuantumChem., 2000, 80: 439.
[18] Ventura O N, Kieninger M, Cachau R E,etal. Density functional computational thermochemistry: determination of the enthalpy of formation of sulfine, CH2SO, at room temperature[J].Chem.Phys.Lett., 2000, 329: 145.
[19] Denis P A, Ventura O N. Density functional investigation of atmospheric sulfur chemistry II. The heat of formation of the XSO2radicals X=H,CH3[J].Chem.Phys.Lett., 2001, 344: 221.
[20] Okamoto Y. Can Ti2-C2H4complex adsorb H2molecules[J].J.Phys.Chem. C, 2008, 112: 17721.
Theoretical study of thermodynamic and dynamical properties of hydrogen absorption on titanium-ethylene complex
HE Na1, GAO Tao2
(1. Institute of Technology, Xianning Vocational Technical College, Xianning 437000, China; 2. Institute of Atomic and Molecular Physics, Sichuan University, Chengdu 610065, China)
Recently, we have predicted the geometries of hydrogen storage in titanium-ethylene complex. Here we further employ density functional theory (DFT) with B3LYP hybrid functional in the calculations of the bond dissociation enthalpy and free energy of these geometries. The conclusions indicate that the reaction C2H4Ti +nH2→C2H4Ti(H2)ncan occur spontaneously and C2H4Ti(H2)ncomplex is stable at 298 K、250 K、200 K. Meanwhile this reaction is exothermic reaction, so the reaction is easier with the decreasing temperature. Besides, we get kinetic curves with Born-Oppenheimer molecular dynamics (BOMD) simulations for C2H4Ti(H2)5complex at 298 K, 250 K and 200 K, respectively. The study on dynamical properties indicates that C2H4Ti(H2)5complex is not stable at 298 K for a long time. While C2H4Ti(H2)5complex is more stable at 200 K, which further indicates that decreasing temperature is conducive to the stability of titanium ethylene molecule hydrogen.
Titanium-ethylene; Thermal correction to enthalpy; Thermal correction to Gibbs free energy; Born-Oppenheimer molecular dynamics
103969/j.issn.1000-0364.2015.12.022
2014-02-25
何娜(1983—),女,湖北人,讲师,主要从事原子与分子物理研究.E-mail: hena23031@aliyun.com.cn
0552.3+3
A
1000-0364(2015)06-1044-06
猜你喜欢
中国特种设备安全(2022年4期)2022-07-08
中国特种设备安全(2022年4期)2022-07-08
无机盐工业(2022年3期)2022-03-11
中学生数理化(高中版.高考理化)(2021年5期)2021-07-16
汽车实用技术(2018年7期)2018-05-18
电子制作(2016年19期)2016-08-24
学园(2015年5期)2015-10-21
汽车与新动力(2014年4期)2014-02-27
海外文摘(2001年1期)2001-04-09
