
使用肺癌GWAS数据进行遗传风险预测的方法和策略研究
2015-03-09 12:56南京医科大学公共卫生学院生物统计学系211166
中国卫生统计 2015年4期
南京医科大学公共卫生学院生物统计学系(211166)
段巍巍 赵 杨 张丽伟 胡志斌 陈 峰△
使用肺癌GWAS数据进行遗传风险预测的方法和策略研究
南京医科大学公共卫生学院生物统计学系(211166)
段巍巍 赵 杨 张丽伟 胡志斌 陈 峰△
目的探讨基于肺癌全基因组关联研究数据的遗传风险预测方法和策略。方法将肺癌GWAS数据中的南京子样本和北京子样本分别作为训练集和测试集,分别使用预测全集和最优预测子集两种策略,比较三种预测方法在不同连锁不平衡结构(LD)和初筛检验水准(α)下的预测准确度。结果wGRS在高LD结构下,随着-log(α)增大,预测准确度呈现上升趋势;RF和SVM对LD结构不如wGRS敏感,但三种方法在低LD结构(r2<0.2)下预测准确度优于高LD结构;wGRS方法下最优预测子集效果略优于预测全集效果,SVM下子集效果与全集近似,但略逊于全集,RF下子集效果则不如全集,且差距较大。结论基于LD结构修剪SNP位点和选择适当的初筛水准可以提高遗传风险预测准确度,此时wGRS方法预测效果优于SVM和RF。
肺癌 遗传风险得分 支持向量机 随机森林 最优预测子集 单核苷酸多态性
近些年来,全基因组关联研究(genome-w ide association study,GWAS)蓬勃发展。截止2013年,全球的研究者们累计发现了与600多种表型(疾病)相关的15000多个单核苷酸多态性(single nucleotide polymorphism,SNP)位点[1],而利用这些已发现的位点进行临床个体化医疗实践、疾病预防等成为后GWAS(post-GWAS)时代的主要目标之一。而建立一个准确的遗传风险预测模型则成为GWAS研究成果转化最为关键的一步。早期的遗传风险预测研究表明,利用GWAS获得的关联位点进行预测并不成功[2-4],其中的一个主要原因在于GWAS研究为了控制假阳性关联位点,多阶段验证过于严格,具体到某一类疾病(表型)而言,关联位点数量较少[5-7]。本研究基于肺癌病例-对照GWAS研究数据,选择灵活的关联位点初筛水准,采用新的策略,即以能否增加预测效果作为进一步纳入SNP位点进行预测的标准,并采用多种预测方法进行肺癌的遗传风险预测效果分析和比较。
资料与方法
1.研究对象与质量控制
数据来自于一项非小细胞肺癌的病例-对照GWAS研究[7],包含南京样本(1473个病例和1962个对照)和北京样本(858个病例和1115个对照)两个部分,详细的样本人群信息参见文献[7]。所有研究个体的DNA样本来自于全血,使用Affymetrix Genome-W ide Human SNP Array 6.0芯片进行基因分型,经过严格的质量控制后[7-8],用于分析的共570373个SNP位点。
2.研究策略与统计分析
(1)研究策略
将研究数据集分为训练集和测试集。首先在训练集中进行位点初筛,即每一个位点都与表型变量做logistic回归。随后仅对P值小于初筛水准α的位点进行分析。对初筛后的位点考虑两种研究策略:①全部纳入初筛时有统计学意义(P<α)的位点进行预测分析,简称“预测全集”,即Performance[phenotype,score(SNP)];其中Performance表示预测准确度,phenotype表示表型变量,连续表型时使用决定系数(R2),二分类表型时使用受试者工作曲线(receiver operating characteristic curve,ROC)下面积(area under curve,AUC);score表示个体在候选位点上的得分值。②仅纳入①中位点的最优预测子集,选择方法如下:
a.对于所有初筛后的m个位点,选择其中具有最佳预测准确度的位点。对于连续表型,有标准化后基因型矩阵G和表型向量y,令相关系数向量corr=Gy则向量中最大值对应的位点即为m个位点中的最佳预测位点,此处为每个位点回归系数组成的向量;同理,对于二分类表型,则选择m个位点中AUC最大的位点。
b.从剩下的m-1个位点中挑选一个位点纳入预测模型,使得新模型的预测能力提升最大。选择第i个位点:表示现有模型的遗传得分表示第i个位点的遗传得分。
c.重复过程b,直到纳入某一个位点后模型的预测能力不再提高时,则停止搜索。此时纳入模型的SNP位点即组成了最优预测子集。
最后,在测试集中评价相关预测方法和上述策略的预测效果。
(2)统计分析
用南京样本数据作为训练集,北京样本数据作为测试集。位点初筛时使用logistic回归,采用相加生物学模式,并校正了如下因素:年龄、性别、吸烟(包/年)以及人群分层的前两个主成分。本文使用的遗传预测方法有:
①效应加权遗传风险得分(effect-weighted genetic risk score,wGRS),即此时Gi表示第i个位点的次等位基因计数(取值为0、1、2)向量,wi为第i个位点的权重,wGRS的方法是现行比较通用的方法[9-12]。
②支持向量机(support vectormachine,SVM),在SVM的参数设置中,考虑到位点初筛后的数据结构(较低的维度和可能的非线性关系)以及常用核函数的相关性质,本文拟选用高斯核函数(radial basis function,RBF),即与此同时,将其惩罚参数Cost设置为1,而核参数Gamma设置为位点数的倒数。SVM的相关原理和遗传风险预测可以参考其他研究[13-15]。
③随机森林(random forest,RF),主要参数中森林中决策树的数量(ntree)、内部节点随机选择的特征数(m try)及终末节点最小样本数(nodesize)分别设置为500、位点数开方和1,RF的原理和分类应用可以参见早期的研究[16-18]。在应用相关预测方法之前我们对缺失数据进行了均值填补。
数据处理和分析使用plink 1.7软件和R v2.15语言包,其中SVM算法使用R语言e1071包,RF算法使用random Forest包。
(3)遗传风险预测评价指标
本研究选择AUC作为预测效果评价指标,其理论取值范围为0.5~1,取值越大,表明预测能力越强。AUC及其方差的估计使用Mann-Whitney检验的U统计量[19]。
(4)研究参数设置
考虑到GWAS研究中多重比较的检验水准(α)较为严格,我们在预测研究中设定了灵活的检验水准,即-log(α)设定为4~9;为了考虑连锁不平衡(linkage disequilibrium,LD)结构对预测结果的影响,我们对候选的SNP位点进行了修剪,分别设定LD参数为r2<1、r2<0.5和r2<0.2,分别表示不修剪任何SNP位点、修剪掉r2大于0.5的位点、r2大于0.2的位点。
结 果
在两种研究策略下分别比较wGRS、SVM和RF三种分类方法的预测能力。表1为wGRS方法下训练集自身验证(self-validation)的结果。结果表明,最优预测子集的预测效果始终是优于全集,并具有较高的AUC值。LD结构是遗传数据分析中不可忽视的影响因素,随着r2阈值的降低,纳入各个预测集合的位点数降低。对于预测全集而言,r2<0.5与r2<0.2情况下预测能力优于r2<1,而它们之间的差距随着初筛阈值逐渐严格(此时组成各自预测全集的位点集逐渐相同)而变小;在相同的α值下,高LD阈值下构建的预测全集包含了低LD下的预测全集位点,而类似情况下,预测子集则不存在这种包含关系。随着-log(α)的逐渐增大,AUC基本呈现一个缓慢下降的趋势。
测试集验证的结果见图1。SVM随着-log(α)增大,预测全集与最优预测子集保持着相同的变化趋势,RF在-log(α)<7时AUC值保持稳定,随后下降明显,而SVM则在5<-log(α)<7时稳定,过高或过低的α值都会引起AUC值的下降;考虑到SVM和RF在共线性问题上相较wGRS更加稳健、SVM对于噪声位点的耐受性[20]以及RF可以纳入位点间可能的交互作用等因素,在较低的-log(α)下,其结果优于wGRS,但随着集合内位点减少,其效果亦出现下降;RF在-log(α)=4处呈现领先优势,可能原因在于交互作用处理上的优势。由于wGRS对高LD的敏感性,其在高LD结构下,随着-log(α)增大,保持着上升趋势(需要指出的是,本研究数据的特征是:-log(α)越大,全集和子集内位点数减少,集合内高相关的位点也逐渐减少);而在低LD结构下,随着-log(α)增大,集合内位点数目成为主要影响因素,因此呈现出与高LD不同的变化趋势。r2<0.2情况下预测结果基本上优于前两者;wGRS方法下预测子集效果略优于预测全集效果,SVM下子集效果与全集近似,但略逊于全集,RF下子集效果则不如全集,且差距较大,可能的原因在于构建最优预测子集时位点采用线性结合方式,因此不适用于SVM和RF。在r2<0.2和-log(α)=6处获得参数组合下最大AUC,估计值为0.7149,95%CI为0.6912~0.7387。

表1 训练集自身验证预测准确度
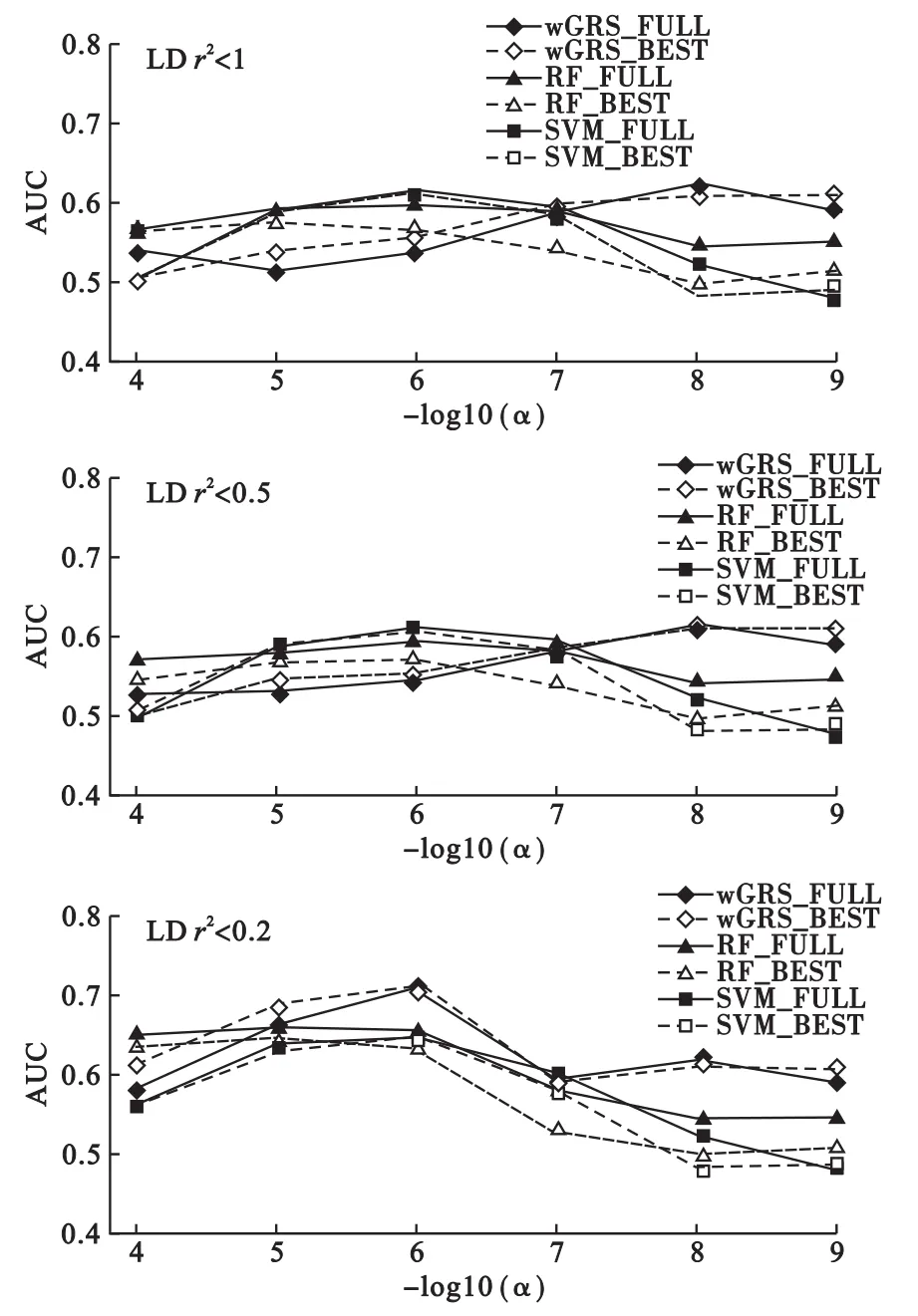
图1 测试集中预测准确度变化图
讨 论
本研究采用预测全集和最优预测子集两种研究策略,在不同LD结构参数和初筛水准α下比较wGRS、SVM和RF这三种常用预测方法的预测准确度。总体而言,最优子集下的wGRS方法在r2<0.2和-log(α)=6的参数下获得了较为满意预测准确度(AUC=0.715)。而对比同类研究,Li等人[12]利用其他GWAS研究发现的4个肺癌关联位点进行预测,其AUC值仅为0.555。Wei等人[13]在1型糖尿病的预测研究中也发现,设定适当宽松的α值有助于提高预测准确度。如果假设SNP位点的效应服从于一个均数为0的正态分布[21],那么大部分的关联研究都仅发现了分布尾巴处"数量少、效应大"的位点。由此可见,忽略中间大部分低效应位点必然影响预测模型的准确度,而宽松的α值可以在一定程度上纳入这些潜在的关联位点,当然本研究和Wei等人的研究也都展示了这种“宽松”是有限度的,即受制于噪声位点的大量增多,预测准确度会逐渐降低。
高LD结构对线性结合的wGRS方法影响较大,因此参照其他研究对SNP位点进行修剪(LD-pruning)[22-24]。RF和SVM对LD结构不如wGRS敏感,但三种方法在低LD结构下表现均优于高LD。对于两种预测位点集合,考虑到训练集与测试集可能存在数据结构上的差异,最优预测子集在测试数据中并不是完全优于预测全集,而构建最优预测子集的方法采用了wGRS的得分相加方式,因此最优子集并不适用于RF和SVM方法。综合而言,在低LD结构的情况下,wGRS方法优势显著,此时可以选择最优预测子集;而在高LD和宽松的初筛阈值下,由于过多的假阳性位点和共线性结构,所有方法均表现不佳,但RF相对稳定。
在同类的遗传风险预测研究中,有相当一部分的结果[25-27]不甚理想,而这些预测研究的共同特点在于使用的风险位点均是经过多阶段严格验证。由此可见,受限于研究设计和相关技术,利用苛刻条件下发现的少量常见变异、强效应关联位点很难获得令人振奋的预测能力。但是我们不应该以消极的态度来看待关联研究的转化应用。值得注意的是,我们认识到遗传度(heritability)低的疾病会限制遗传预测方法效果的提升,以本研究的肺癌疾病为例,国外的一项研究表明[28],瑞典人群肺癌的遗传度仅在8%左右。对于本研究中使用的预测指标AUC,很多学者提出异议[4,29],他们发现数个较强的预测指标线性组合后的AUC值相较某单一指标提高幅度有限,于是对AUC作为预测评价指标产生了质疑,并提出了再分类优值(net reclassification improvement,NRI)、综合判别优值(integrated discrimination improvement,IDI)等指标[29],但考虑到AUC指标的综合评价能力、广泛的通用性以及研究目的,本文依然选择AUC作为研究的预测评价指标。这里讨论的方法均为成熟或经典的预测方法,最近有研究者[30-31]在探讨使用混合效应模型进行预测,亦有研究者整合多组学信息进行预测研究[32],但这些方法还有待大量的实例研究去验证。
(致谢:感谢南京医科大学公共卫生学院分子流行病实验室和北京子研究的老师、同学给予的支持)
[1]Welter D,MacArthur J,Morales J,et al.The NHGRIGWAS Catalog,a curated resource of SNP-trait associations.Nucleic Acids Res,2014,42(Database issue):D1001-1006.
[2]van der Net JB,Janssens AC,Sijbrands EJ,et al.Value of genetic profiling for the prediction of coronary heart disease.Am Heart J,2009,158(1):105-110.
[3]M ihaescu R,Meigs J,Sijbrands E,etal.Genetic risk profiling for prediction of type2 diabetes.PLoSCurr,2011,3:RRN1208.
[4]Cook NR.Use and misuse of the receiver operating characteristic curve in risk prediction.Circulation,2007,115(7):928-935.
[5]McCarthy MI,Abecasis GR,Cardon LR,etal.Genome-wide association studies for complex traits:consensus,uncertainty and challenges.Nat Rev Genet,2008,9(5):356-369.
[6]Manolio TA.Genomewide association studies and assessment of the risk of disease.N Engl JMed,2010,363(2):166-176.
[7]Hu Z,Wu C,Shi Y,etal.A genome-w ide association study identifies two new lung cancer susceptibility loci at13q12.12 and 22q12.2 in Han Chinese.Nature genetics,2011,43(8):792-796.
[8]陈峰,柏建岭,赵杨,等.全基因组关联研究中的统计分析方法.中华流行病学杂志,2011,32(4):400-404.
[9]Speliotes EK,W iller CJ,Berndt SI,et al.Association analyses of 249,796 individuals reveal18 new lociassociated with body mass index.Nat Genet,2010,42(11):937-948.
[10]Ripatti S,Tikkanen E,Orho-Melander M,et al.A multilocus genetic risk score for coronary heart disease:case-control and prospective cohort analyses.Lancet,2010,376(9750):1393-1400.
[11]Gui L,Wu F,Han X,et al.A multilocus genetic risk score predicts coronary heart disease risk in a Chinese Han population.Atherosclerosis,2014,237(2):480-485.
[12]Li H,Yang L,Zhao X,et al.Prediction of lung cancer risk in a Chinese population using amultifactorial geneticmodel.BMCMed Genet,2012,13:118.
[13]Wei Z,Wang K,Qu HQ,et al.From disease association to risk assessment:an optim istic view from genome-w ide association studies on type 1 diabetes.PLoSGenet,2009,5(10):e1000678.
[14]Becker N,Toedt G,Lichter P,et al.Elastic SCAD as a novel penalization method for SVM classification tasks in high-dimensional data.BMC Bioinformatics,2011,12:138.
[15]Cortes C,Vapnik V.Support-vector networks.Machine learning,1995,20(3):273-297.
[16]Breiman L.Random forests.Machine learning,2001,45(1):5-32.
[17]Zhang H,Yu CY,Singer B.Cell and tumor classification using gene expression data:construction of forests.Proc Natl Acad Sci USA,2003,100(7):4168-4172.
[18]Yoon D,Kim YJ,Park T.Phenotype prediction from genome-w ide association studies:application to smoking behaviors.BMC Syst Biol,2012,6(2):S11.
[19]Mason SJ,Graham NE.Areas beneath the relative operating characteristics(ROC)and relative operating levels(ROL)curves:Statistical significance and interpretation.Quarterly Journal of the Royal Meteorological Society,2002,128(584):2145-2166.
[20]Xing EP,Jordan MI,Karp RM.Feature selection for high-dimensional genom ic m icroarray data.in ICML,2001:Citeseer.
[21]Speed D,Hemani G,Johnson MR,et al.Improved heritability estimation from genome-w ide SNPs.Am JHum Genet,2012,91(6):1011-1021.
[22]Purcell SM,W ray NR,Stone JL,et al.Common polygenic variation contributes to risk of schizophrenia and bipolar disorder.Nature,2009,460(7256):748-752.
[23]Consortium SWGO.Biological insights from 108 schizophrenia-associated genetic loci.Nature,2014,511(7510):421-427.
[24]Chatterjee N,Wheeler B,Sampson J,et al.Projecting the performance of risk prediction based on polygenic analyses of genome-w ide association studies.Nat Genet,2013,45(4):400-5,405e1-3.
[25]Zheng SL,Sun J,Wiklund F,etal.Genetic variantsand family history predict prostate cancer sim ilar to prostate-specific antigen.Clin Cancer Res,2009,15(3):1105-1111.
[26]Mealiffe ME,Stokowski RP,Rhees BK,et al.Assessment of clinical validity of a breast cancer risk model combining genetic and clinical information.JNatl Cancer Inst,2010,102(21):1618-1627.
[27]Wacholder S,Hartge P,Prentice R,et al.Performance of common genetic variants in breast-cancer risk models.N Engl JMed,2010,362(11):986-993.
[28]Czene K,Lichtenstein P,Hemminki K.Environmental and heritable causes of cancer among 9.6million individuals in the Swedish family -cancer database.International Journal of Cancer,2002,99(2):260-266.
[29]Pencina MJ,D’Agostino RS,D’Agostino RJ,et al.Evaluating the added predictive ability of a new marker:from area under the ROC curve to reclassification and beyond.StatMed,2008,27(2):157-72;discussion 207-212.
[30]Golan D,Rosset S.Effective genetic-risk prediction using mixed models.Am JHum Genet,2014,95(4):383-393.
[31]Speed D,Balding DJ.MultiBLUP:improved SNP-based prediction for complex traits.Genome Res,2014,24(9):1550-1557.
[32]Wheeler HE,Aquino-M ichaels K,Gamazon ER,et al.Poly-om ic prediction of complex traits:OmicKriging.Genet Epidemiol,2014,38(5):402-415.
(责任编辑:郭海强)
Strategies of Genetic Risk Prediction with Lung Cancer GWASData
Duan Weiwei,Zhao Yang,Zhang Liwei,et al.(DepartmentofBiostatistics,NanjingMedicalUniversity(211166),Nanjing)
ObjectiveTo investigate the performance of three genetic risk prediction methods,weighted genetic risk score(wGRS),supportvector machine(SVM)and random forest(RF),applied to high dimensional data of lung cancerwith two strategies.MethodsThis study served Nanjing and Beijing samples of GWAS data as training set and testing set respectively.Wemade use of the two strategies of Full predictive subset(FS)and Best predictive subset(BS)and compared the prediction accuracy within the threemethodsmentioned above with the combination of Linkage Disequilibrium(LD)and hypothesis testing levels(α).ResultsUnder a high LD structure,the prediction accuracy of wGRSwas on the rise with the increasing-log(α).RF and SVM were not sensitive to LD structures as wGRS,but the predictive accuracy of each method applied with a low LD structure(r2<0.2)wasmainly better than itself with a high LD structure.Moreover,the performance of BS was slightly better than,approximately equal to or tiny less than and worse than FSwhen themethodswere respectively wGRS,SVM and RF.ConclusionThe prediction accuracy could be improved with the condition of LD-pruning and adopting a properα-value,meanwhile,wGRSwas better than SVM and RF in that condition.
Lung cancer;Genetic risk score;Support vectormachine;Random forest;Best predictive subset;Single nucleotide polymorphism
国家自然科学基金(81473070,81373102)
△通信作者:陈峰,Email:fengchen@njmu.edu.cn
猜你喜欢
安庆师范大学学报(自然科学版)(2021年1期)2021-11-28
昆明医科大学学报(2021年5期)2021-07-22
中华养生保健(2020年3期)2020-11-16
合肥工业大学学报(自然科学版)(2020年2期)2020-03-23
南京大学学报(数学半年刊)(2020年1期)2020-03-19
World Journal of Clinical Cases(2019年6期)2019-04-17
艺术评论(2017年12期)2017-03-25
中国卫生标准管理(2015年3期)2016-01-14
都市丽人(2015年4期)2015-03-20
民主与科学(2014年5期)2014-02-28
