
黑蜂王台病毒基因组结构研究进展
2015-03-08 09:35:26宋战昀石建平崔焕忠王振国孟庆峰王全凯李敏思王向辉
中国预防兽医学报 2015年1期
杨 倩,宋战昀,张 健,石建平,崔焕忠*,王振国,孟庆峰,王全凯,4,李敏思,郑 言,王向辉
(1.吉林农业大学,吉林 长春 130118;2.吉林出入境检验检疫局,吉林 长春 130062;3.长春生物制品研究所有限责任公司,吉林 长春 130062;4.吉林省中韩动物科学研究院,吉林 长春 130600;5.甘肃农业大学,甘肃 兰州 730070)
蜜蜂病毒被认为是主要威胁蜜蜂健康的因素之一,是引发蜜蜂蜂群大量死亡及蜂群衰竭的关键因素[1-7]。首次鉴定出蜜蜂病毒并认定其为新的感染蜜蜂的病原体是在20 世纪初期。目前,已知可感染蜜蜂的病毒高达20 种,其中包括18 种RNA 病毒,其中的某些病毒已在全球范围内广泛传播[8-10]。蜜蜂病毒能够影响蜜蜂形态、生理和行为,并与弱群及蜜蜂死亡息息相关[11]。蜜蜂黑蜂王台病是由黑蜂王台病毒(Black queen cell virus,BQCV)引起的一种蜜蜂蜂王幼虫病害。BQCV 由Baliey 等于1977 首次报道,该病毒是从蜂王幼虫和封盖蜂王蛹中分离得到,感染的蜂王蛹死亡并且其颜色由棕色变暗变黑,随后蜂房壁开始变黑,BQCV 的名称也因此而来[12]。最新研究发现,BQCV 能够在世界范围内的不同蜜蜂种间甚至是非膜翅类昆虫种属间广泛传播,在西方国家(尤其是中欧国家)及西方蜜蜂蜂群中,BQCV 是所有蜜蜂病毒中流行最广的一种病毒[13-16]。它通常以一种无病症的隐性感染方式长期存在于被感染蜂群的成蜂及幼虫体内,表面健康的蜂群可能隐藏病毒,BQCV 主要呈慢性感染。这种感染方式是不易观察到的,直到蜂王幼虫及蛹开始变黑,最终导致整个巢房房壁变黑,才呈现出可观察的现象,但此时已导致蜂群的大量死亡,造成无法挽回的损失。本文主要从BQCV 基因组结构进行综述。
1 BQCV的基本病毒学特征
2000 年,Leat 曾将BQCV 归类于类微小核糖核酸病毒(PicoRNA-like viruses)[17]。2002 年,Mayo 将BQCV 划分到双顺反子病毒科(Dicistroviridae)[18],蟋蟀麻痹病毒属(Cripavirus),这个分类一直沿用至现在。最初发现在死亡的蛹内有大量等轴的病毒粒子,直径为30 nm,无囊膜,呈正二十面体球型结构。病毒粒子中包含有一个单股正链RNA,编码4 种衣壳蛋白,并由60 个VPl、VP2 和VP3 3种蛋白构成衣壳,还有一个小的蛋白VP4。这4 种蛋白分子量分别为34 ku、32 ku、29 ku 和6 ku。该病毒的单股正链RNA 可以直接作为模板mRNA,进行病毒蛋白的翻译。病毒的沉降系数为151 S,浮密度为1.34 g/mL(CsCl)[17]。
2 BQCV的分类地位
关于病毒-机体的相互作用,感染昆虫的病毒与感染脊椎动物的病毒有很大差异。在感染脊椎动物的病毒中,中和抗体对病毒粒子表面抗原具有强选择性。因此,通常可观察到相同病毒不同病毒株的编码结构蛋白的基因组节点的抗原决定簇上存在序列多样性。而在感染昆虫的病毒中,免疫系统不产生免疫球蛋白,防御机制主要是依靠固有免疫[19-20]。因此,昆虫病毒的进化可能受到以病毒基因组不同区域为目标的其他细胞内因素的影响。很多情况下,BQCV 的感染是不明显的,但却能够导致蜂房内蜂蛹的大量死亡。除了宿主因素和养蜂条件外,BQCV 不同病毒株的毒力差异也能影响该病的发展。
2000 年,Leat 等分离到BQCV South Afica(南非)株(AF 183905)并进行了全基因组测序[17],该病毒具有类小核糖核酸病毒的生物学特性。当初Leat 等确定该病毒分类的基础是昆虫感染的RNA 病毒,并且是类小核糖核酸病毒,其代表病毒包括果蝇属C 病毒(Drosophila C virus,DCV),禾谷益管蚜病毒(Rhopalosiphum padivirus,RhPV)、褐飞虱双顺反子病毒(Himetobi P virus,HiPV)和斯氏珀蝽病毒(Plautia staliintestine virus,PSIV),而且已获得这几种病毒的全基因组序列。将其分离得到的BQCV 南非株与这4 种病毒对比,发现相同之处:均属于双顺反子病毒,并且均具有一个5' 端ORF 编码的复制酶蛋白和一个3' 端ORF 编码衣壳蛋白。进一步发现,它们的RNA 依赖的RNA 聚合酶(RNA dependent RNA polymerase,RdRp)高度相似,最终确定该BQCV 的分类地位为类微小核糖核酸病毒[17]。Mayo 将BQCV 划分到双顺反子病毒科,蟋蟀麻痹病毒属[18]。Donaldson 在2010 年研究人畜共患病时,研究感染蝙蝠样本的昆虫病毒时发现,BQCV 可感染蝙蝠[21],并与蜜蜂克什米尔病毒(Kashmir bee virus,KBV)、蟋蟀麻痹病毒(Cricket paralysis virus,CrPV)和蚜虫致死麻痹病毒(Aphid lethal paralysis virus,ALPV)共同归类于双顺反子病毒科,进一步证实该分类的准确性。
3 BQCV的全基因组序列分析
关于BQCV 全基因组序列的研究,自Leat 于2000 年对南非株首次测序之后,GenBank 中登录的BQCV 全基因组序列截止到目前共有6 株。Leat 研究显示,BQCV 南非株核苷酸序列全长为8 550 nt,不包括多聚A 尾(图1)。基因组包含了高比例的A 和U 核苷酸,由29.2 % A,30.6 % U,18.5 % C 和21.6 % G 组成[5]。基因组包括两个不重叠的ORF1 和ORF2,两个ORF 之间存在一非编码的插入序列(IGR)。在5' 端非编码区(UTR)和IGR 区均有核糖体结合位点(IRES),具有启动病毒蛋白的翻译功能,但两个IRES 序列及其翻译启动机制不同。Tapaszti 等发现的4 株中欧基因型[22],分别是:BQCV Hungary-10(8 335 nt)、BQCV Poland-4(8 333 nt)、BQCV Poland-5(8 298 nt)和BQCV Poland-6(8 302 nt)、2013 年Reddy 等发现的BQCV South Korea(8 392 nt)[23]。BQCV 的ORF1编码解旋酶(Helicase),3C 样半胱氨酸蛋白酶(Protease)和RdRp。ORF2 的翻译起始是通过IRES 完成的,翻译的起始密码子为CCU,可以促进ORF2 的翻译起始,编码4种衣壳蛋白。
Reddy 将韩国株与中欧基因型及南非株做进化树分析(图2)[23],结果显示:该株与匈牙利-10 具有92 %的同源性,与南非株同源性较低(86 %),与波兰-4 为89 %的相似性,与波兰-5 和波兰-6 均为91 %的相似性。
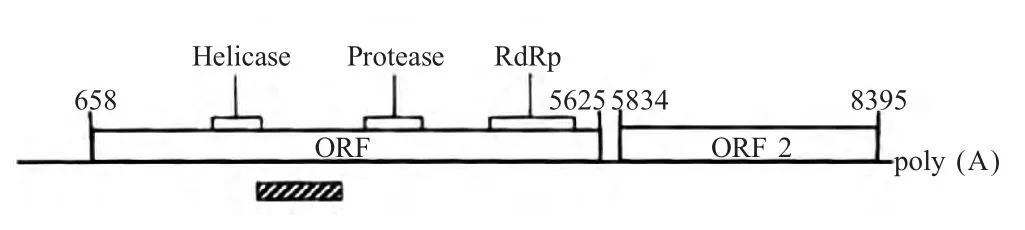
图1 BQCV(南非参考株)基因组示意图
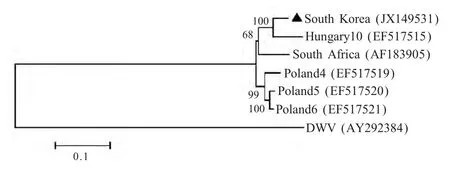
图2 韩国BQCV 分离株与其他分离株的全基因组系统进化分析(以蜜蜂卷翅病毒(DWV 全基因组序列作为外群)
4 BQCV的基因组结构分析
4.1 ORF1 ORF1 的长度测定显示,南非株为4 968 nt(658 nt~5 625 nt),中欧的4 株基因型的ORF1 的长度均为4 887 nt,其中匈牙利-10 和波兰-4 两株的核苷酸序列位置为605 nt~5 491 nt,波兰-5 和波兰-6 两株的位置为606 nt~5 492 nt,而韩国株为4 890 nt(591 nt~5 480 nt)[17,22-23]。在南非株[17,22]中发现,在658 nt~660 nt 之间存在一个起始密码子AUG,在5 623 nt~5 625 nt 之间有一个终止密码子UAG。这些密码子代表了ORF1 的第一个可能的起始密码子和终止密码子。在南非株核苷酸序列位置为1 177 nt~1 245 nt 区域间是一段长为69 nt 的连续片段,翻译成氨基酸序列为EAHCKIKSFCTLFKCSRHFNNVQ,而该段序列在韩国株和中欧基因型中均是不存在的[23]。Tapaszti 认为ORF1 具有较高的序列多样性,能够将它分成两个不同的区域[22]:置换区域和插入/缺失区域。在ORF 1 靠近5' 端的位置,一段大约长为1 700 nt的区域,中欧型的该段序列比南非株短84 nt。在ORF1的3' 端,大约在1 700 nt~5 625 nt,中欧型与南非株比较,该段序列的相似度要高于前面所述的1 700 nt 长的序列,并且符合病毒的地理起源[22]。该ORF 1 区域的氨基酸序列与非结构蛋白的氨基酸序列极为相似,推断其编码复制酶蛋白,由1 655 个氨基酸组成,计算其分子量为189 471 ku。推断ORF1 区域可编码3 种非结构蛋白,分别为:Helicase、Protease 和RdRp 3 种酶,它们在氨基酸序列上的起始位置分别为aa441、aa902 和aa1 317。1)编码 Helicase 区 域:在 Helicase 氨基酸序列的452GXXGXGKS459和510QX5DD517高度保守[17]。据Tapaszti 等研究,在该区域,中欧基因型显示出与南非参考株具有82 %~90 % 的一致性[22]。其 中,匈牙利株(EF517506-EF517515)与奥地利株(EF517501-EF517505)彼此间具有更高的一致性(94 %~99 %),与波兰株(EF517516-EF517522)仅有81 %~96 %的一致性。波兰株-4、5、6(EF517519-EF517521)非常相近,一致性达到98%~99 %,并且这3 株相比于其他中欧基因型(81 %~83%),与南非株(90 %)更相近。Helicase 由186 个氨基酸组成,将其推导氨基酸序列在中欧各基因型之间相互比较,并同南非株比较,该氨基酸序列可达91 %~100 %的一致性。2)编码Protease 区域:在保守区域1056GXCG1059中的组氨酸(H909)、天冬氨酸(D964)(或谷氨酸)和半胱氨酸(C1058),对于蛋白酶的活性而言是保守的也是必要的。3)编码RdRp 区域:通过对比昆虫为传播媒介的动物病毒和植物病毒的RdRp 确定BQCV 的分类地位[17],这表明RdRp 为BQCV 编码的重要蛋白。在氨基酸序列上的aa1 317~aa1 584 之间共有8 个保守区域。
4.2 ORF2 ORF2 编码853 个氨基酸,分子量为95 713 ku。根据它们与衣壳多聚蛋白氨基末端的亲近性将它们归类为CP1、CP2、CP3 和CP4。埃德曼降解法确定CP2-CP4 的氨基末端序列及ORF2 编码的氨基酸序列位置分别为:232AGLKVQPP240、307SKPLLPITN315和574SNSGT578。CP1~CP4 蛋白在由ORF2 编码的氨基酸序列上这4 种蛋白是连续的,可将它们分别切割成:231、75、267 和280 个氨基酸大小。这4 种蛋白的分子量大小分别为:32 ku、6 ku、29 ku 和34 ku。并确定CP1 对应VP2,CP2 对应VP4,CP3 对应VP3 及CP4 对应VP1[5]。在结构蛋白编码区域,中欧基因型与南非基因型间有91 %~94 %的序列一致性。在该区域,匈牙利株与奥地利株再一次表现出高的相似度(97 %~99 %),与波兰株的相似度为93 %~96 %。波兰-4 显示出一个明显的分歧:与其他波兰株、奥地利株和匈牙利株之间有89 %~91 %的相似性,与南非株也有91 %的相似性。将核苷酸序列翻译成氨基酸序列,该区域可以翻译成132 个氨基酸,将中欧各基因型之间相互比较,并同南非株比较,该氨基酸序列一致性可达97 %~100 %[22],Noh 在分析BQCV 韩国株的ORF2 时发现,除Korean Am str3(JQ434131)外,其余韩国株(JQ434127-JQ434136)均表现出极高的亲缘关系,而该株则更接近于中欧基因型和南非参考株[24]。
4.3 其他非编码区域 在BQCV 全基因组中,除两个大的ORFs 为编码区域外,还存在一些非编码区域,包括:IGR、5'UTR、3'UTR 和一个小的基因编码连接蛋白VPg蛋白。1)基因间隔区(IGR):在南非株中,两个ORFs 之间被一个208 nt(5 626 nt~5 833 nt)长的基因间隔区隔开,推断可将其作为IRES,促进BQCV 的ORF2 的翻译起始,起始密码子是CUU(5 834 nt~5 836 nt)[17]。在欧洲型最终调查的5 株病毒(波兰-4、5、6,匈牙利-10,奥地利-5)及韩国株中,在靠近3' 端有一段长为155 nt(5 679 nt~5 833 nt)的片段与南非株的IGR 具有100 %的序列一致性,而这段序列一直延续到ORF2 的前56 个核苷酸处[22-23]。在韩国株中,该端区域的长为316 nt,位于5 480 nt~5 796 nt之间[23]。2)5' 端UTR:南非株的5'UTR 长为657 nt,呈四叶苜蓿形的二级结构,推测可能与蛋白翻译有关。Tapaszti 等在该区域识别出一段长为167 nt(69 nt~235 nt)的片段,6 株病毒(波兰-4、5、6,匈牙利-10,奥地利-5和南非株)100 %相同,而整个5'-UTR 区域的序列一致性为94 %~99 %[22],表明该段序列高度保守。3)3' 端UTR:南非株的3'UTR 为155 nt,3' 端polyA 序列在不同的病毒长度不同,但具有遗传稳定性及防止RNA 分子降解的功能,3' 端序列能折叠成茎环结构。推测可能与RNA 复制有关。4)VPg 蛋白:在基因组5' 端有一个小的基因编码连接蛋白VPg,VPg 蛋白具有稳定基因组5' 端结构、启动翻译和复制功能。
5 小 结
本文对GenBank 中登录的全部6 株BQCV 全基因组序列基因组结构分析进行综述。其中,BQCV(韩国株)与其他地理来源的BQCV 株在编码衣壳蛋白区域存在很近的亲缘关系,但全基因组序列存在差异,尽管ORF1 和ORF2 区域与南非参考株有较高的相似度,但其他非编码区域与中欧基因型有更高的相似度,进而推断出该BQCV(韩国株)可能是欧洲基因型的变种。随后,对这6 株病毒株作更详细的综述其编码区和非编码区。编码区又分为编码结构蛋白区域和编码非结构蛋白区域。而6 株全基因组序列及GenBank 中登录的片段序列得出的编码区域的核苷酸序列和氨基酸序列存在相似性和差异性,即相对保守区域相似和可变区域存在差异较大。通过对BQCV 全基因组序列的综述,为进一步深入研究BQCV 的理化特性及致病性等研究奠定分子基础。
[1]Cox-Foster D L,Conlan S,Holmes E C,et al.A metagenomic survey of microbes in honey bee colony collapse disorder[J].Science,2007,318:283-287.
[2]Van Gngelsdorp D,Evans J D,Saegerman C,et al.Colony collapse disorder:A descriptive study[J].PLoS One,2009,4(8):1-17.
[3]Vangngelsdorp D,Meixner M D.A historical review of managed honeybee populations in Europe and the United States and the factors that may affect them[J].J Invertebr Pathol,2010,103:S80-S95.
[4]Ratnieks F L,Carreck N L.Clarity on honey bee collapse?[J].Science,2010,327:152-153.
[5]Carreck N L,Ball B V,Martin S J.Honey bee colony collapse and changes in viral prevalence associated with Varroa destructor[J].J Apic Res,2010,49:93-94.
[6]Higes M,Martín-Herna'ndez R,Botías C.How natural infection by Nosema ceranae causes honey bee colony collapse[J].Environ Microbiol,2008,10:2659-2669.
[7]Higes M,Martín-Herna'ndez R,Garrido-Bailón E,et al.Honey bee colony collapse to Nosema ceranae in professional apiaries[J].Environ Microbiol,2009,1:110-113.
[8]Allen M F,Ball B V.The incidence and world distribution of honey bee viruses[J].Bee World,1996,77:141-162.
[9]Bromenshenk J J,Henderson C B,Wick C H,et al.Iridovirus and microsporidian linked to honey bee colony decline[J].PLoS One,2010,5(10):e13181.
[10]de Miranda J R,Dainat B,Locke B,et al.Genetic characterisation of slow bee paralysis virus of the honeybee(Apis mellifera L.)[J].J Gen Virol,2010,91(10):2524-2530.
[11]Genersch E,Aubert M.Emerging and re-emerging viruses of the honey bee(Apis mellifera L.)[J].Vet Res,2010,41(6):54-59.
[12]Bailey L,Woods R D.Two more small RNA viruses from honey bees and further observations on sacbrood and acute bee-paralysis viruses[J].J Gen Virol,1977,37:175-182.
[13]Tentcheva D,Gauthier L,Zappulla N,et al.Prevalence and seasonal variations of six bee viruses in Apis mellifera L.and Varroa destructor mite populations in France[J].Appl Environ Microbiol,2004,32:7185-7191.
[14]Berényi O,Bakonyi T,Derakhshifar I,et al.Occurrence of six honeybee viruses in diseased Austrian Apiaries[J].Appl Environ Microbiol,2006,34:2414-2420.
[15]Baker A,Schroeder D.Occurrence and genetic analysis of picorna-like viruses infecting worker bees of Apis mellifera L.populations in Devon,South West England[J].J Invert Path,2008,98:239-242.
[16]Kajobe R,Marris G,Budge G,et al.First molecular detection of a viral pathogen in Ugandan honey bees[J].J Invertebr Pathol,2010,104(2):153-156.
[17]Leat N,Ball B,Govan V,et al.Analysis of the complete genome sequence of black queen-cell virus,a picorna-like virus of honey bees[J].J Gen Virol,2000,81:2111-2119.
[18]Mayo M A.Virus taxonomy-Houston[J].Arch Virol,2002,147:1071-1076.
[19]Cherry S,Silverman N.Host-pathogen interactions in Drosophila:new tricks from an old friend[J].Nat Immunol,2006,7:911-917.
[20]Zambon R A,Nandakumar M,Vakharia V N,et al.The Toll pathway is important for an antiviral response in Drosophila[J].PNAS USA,2005,102:7257-7262.
[21]Donaldson E F,Haskew A N,Gates J E,et al.Metagenomic analysis of the viromes of three North American bat species:Viral diversity among different bat species that share a common habitat[J].J Virol,2010,91:13004-13018.
[22]Tapaszti Z,Forgách P,Kovágó C,et al.Genetic analysis and phylogenetic comparison of black queen cell virus genotypes[J].Vet Microbiol,2009,139:227-234.
[23]Reddy K E,Noh J H,Choe S E,et al.Analysis of the complete genome sequence and capsid region of black queen cell viruses from infected honeybees(Apis mellifera)in Korea[J].Virus Genes,2013,47(1):126-132.
[24]Noh J H,Reddy K E,Choe S E,et al.Phylogenetic analysis of black queen cell virus genotypes in South Korea[J].Virus Genes,2013,46(2):362-368.
猜你喜欢
疯狂英语·初中天地(2019年7期)2019-07-23 01:07:02
小天使·一年级语数英综合(2017年9期)2017-10-20 21:12:02
知识经济·中国直销(2016年5期)2016-11-07 09:35:05
阅读与作文(小学高年级版)(2016年5期)2016-05-10 23:32:23
阅读与作文(小学低年级版)(2016年1期)2016-03-12 22:20:48
小朋友·快乐手工(2015年11期)2016-01-07 00:18:50
海外星云(2015年15期)2015-12-01 04:17:40
现代检验医学杂志(2015年6期)2015-02-06 01:44:02
实验动物与比较医学(2014年5期)2014-02-28 14:53:10
中国糖料(2013年1期)2013-01-22 12:28:23
