
药用辅料HFA-134a国家标准纯度分析方法研究
2015-03-03 08:10陈彩琴刘红秀
有机氟工业 2015年3期
陈彩琴 刘红秀 戴 孟
(浙江衢化氟化学有限公司质检中心,浙江 衢州 324004)
药用辅料HFA-134a国家标准纯度分析方法研究
陈彩琴 刘红秀 戴 孟
(浙江衢化氟化学有限公司质检中心,浙江 衢州 324004)
研究了药用辅料HFA-134a国家标准纯度分析方法,分析了标准操作条件的不足之处,优化了气相色谱操作条件,考察了该方法的检出限、线性回收率和精密度,结果证明优化后的方法具有较好的分离度和较高的准确率。
药用辅料HFA-134a;国家标准;色谱条件;进样量
0 前言
四氟乙烷,化学名为1,1,1,2-四氟乙烷,英文名为1,1,1,2-Tetrafluoroethane,分子式C2H2F4,分子质量102.3,CAS号811-97-2,熔点-101 ℃,沸点-26.2 ℃,相对密度1.206(298.15 K,液体)。水中溶解度(298.15 K,101.3×103Pa)为0.15%。四氟乙烷在常温下是一种无色、有轻微醚类气味的气体,一般情况下均压缩成液态储存于钢瓶中。该产品作为药用辅料简称为HFA-134a,作为制冷剂简称为HFC-134a。
国家药品标准四氟乙烷WS5-FG-003-2013于2014年2月28日正式实施,但在实施过程中发现测定纯度的气相色谱条件有些缺陷,如柱温低、出峰慢,主峰拖尾,进样量大,杂质峰被主峰覆盖,分析时间长,实施起来有困难。因此,对国家标准方法进行了优化和验证。采用优化后的国家药品标准测定四氟乙烷主含量和有关物质,分离度更高,峰形美观,分析时间更短,主含量的线性相关系数为0.999 4,有关杂质的线性相关系数均大于0.997 0,回收率为96.3%~102.5%,精密度标准偏差小。该方法能满足药用辅料检测的需要。
1 实验部分
1.1 试剂和材料
药用辅料HFA-134a;配制杂质用纯物质:二氟乙烯(HFC-1132)、五氟氯乙烷(HCFC -115)、三氟甲烷(HFC-23)、1,1,1-三氟乙烷(HFC-143a)、四氟二氯乙烷(HCFC-114/114a)、五氟乙烷(HFC-125)、四氟氯乙烷(HCFC-124)、三氟氯乙烷(HCFC-133a)、三氟二氯乙烷(HCFC-123)。
1.2 仪器和设备
6890气相色谱仪,配PLOT Al2O3色谱柱(50 m×0.53 mm×15 μm),氢火焰离子化检测器。
1.3 气相色谱条件
气化室温度170 ℃,检测器温度250 ℃,柱箱温度80 ℃,保持10 min,以10 ℃/min升温至150 ℃,保持5 min,柱流量2.0 mL/min,分流比5 ∶1。
1.4 试验方法
按要求调试好气相色谱条件,等仪器达到设定状态后,取0.2 mL样品注入气相色谱仪进行分析,分析结束后,仪器自动显示分析报告。
2 结果与讨论
2.1 色谱柱的选择
按四氟乙烷国家药品标准中规定,选取氧化铝为固定相(PLOT Al2O3)的50 m×0.53 mm×15 μm石英毛细管色谱柱为试验对象。
2.2 色谱操作条件的选择
2.2.1 国家药品标准规定的操作条件验证试验
根据选定的气相色谱柱,按国家药品标准规定的操作条件,补充了缺失的柱流量8 mL/min,按照国标操作条件:进样器温度170 ℃,恒流模式,载气流量2.0 mL/min,分流比为5 ∶1,柱温初始温度50 ℃保持2 min,以2 ℃/min从50 ℃升温至100 ℃,再以10 ℃/min从100 ℃升温至150 ℃保持15 min,检测器 (FID)温度250 ℃,氢气流量30 mL/min,进样量为1.0 mL。配制一个混合样,在此操作条件下进行试验,试验结果见图1。

图1 四氟乙烷色谱图(国家药品标准推荐的操作条件)
由于进样量大,柱温条件不合适,主峰峰宽过宽,部分杂质被主峰覆盖,不能有效分离,整个分析过程需要50 min,分析时间长,因此需要重新优化气相色谱条件。
2.2.2 柱温的优化
采用同一样品,在不同柱温条件下按常规气体进样量为0.2 mL进行试验,试验结果见图2~图5。
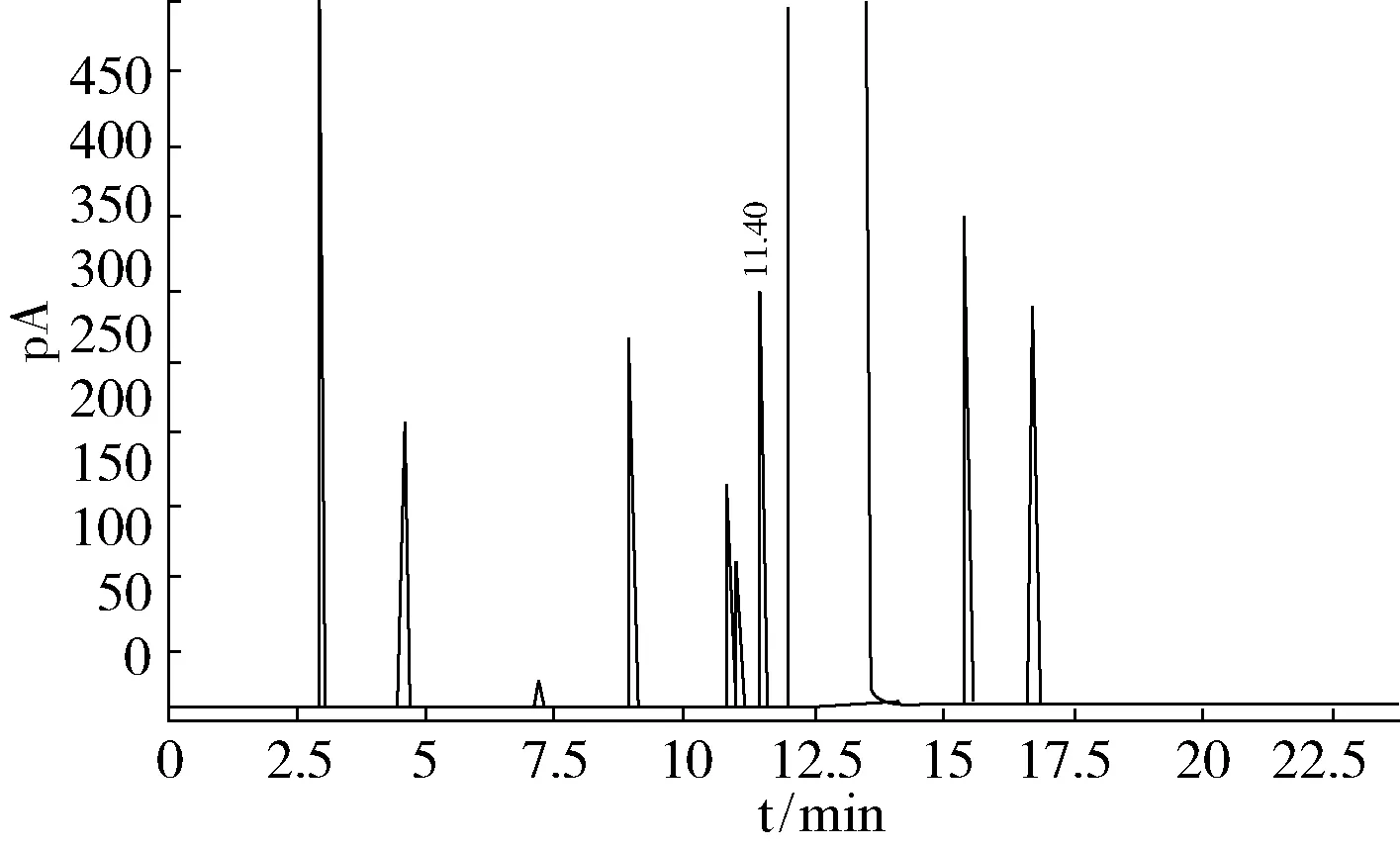
图2 50 ℃温度保持10 min,以10 ℃/min升温至150 ℃,保持5 min

图3 60 ℃温度保持10 min,以10 ℃/min升温至150 ℃,保持5 min

图4 80 ℃温度保持10 min,以10 ℃/min升温至150 ℃,保持5 min
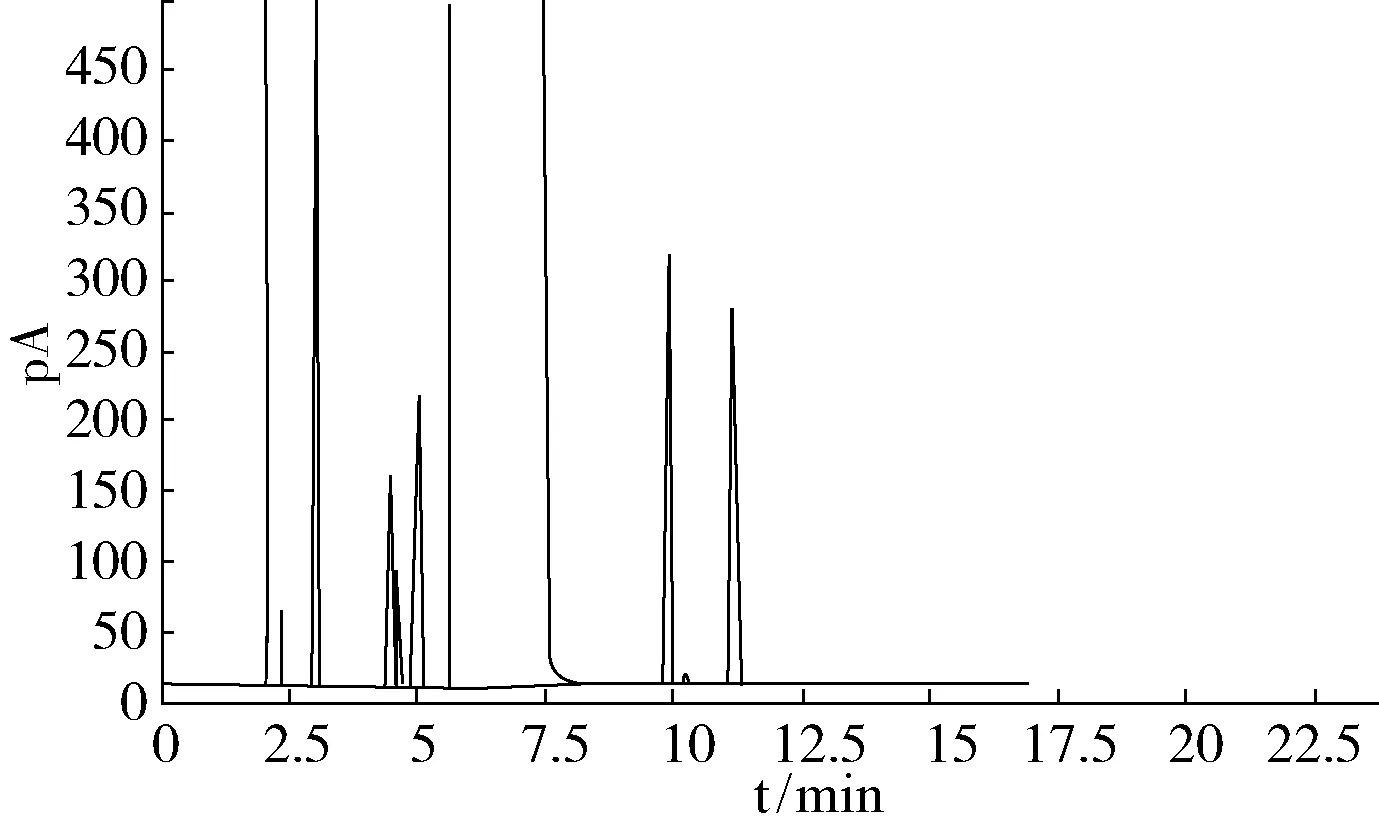
图5 100 ℃温度保持10 min,以10 ℃/min升温至150 ℃,保持5 min
通过比较,图2~图4的操作条件均能使杂质较好地分离,图5出峰过快,杂质含量高时峰不易分开,综合分离效果以及分析时间等因素,最终确定了图4作为最佳操作条件:进样口温度170 ℃、检测室温度250 ℃、柱流量2 mL/min、分流比5 ∶1,柱温初始温度80 ℃保持10 min,以10 ℃/min从80 ℃升温至150 ℃,保持5 min。测定一个样品的时间由50 min缩短至22 min。在此条件下,最难分离的物质对1,1,2,2-四氟-1,2-二氯乙烷和1,1,1,2-四氟-2,2-二氯乙烷的分离度为3.2,五氟乙烷和四氟乙烷的分离度为1.16,两峰完全分离。
2.2.3 进样量的选择
选取0.05 mL、0.1 mL、0.2 mL、0.3 mL、0.4 mL、0.5 mL、0.8 mL、1.0 mL不同的进样量,按优化后的操作条件,分别对同一样品进行试验,结果表明:进样量在0.05~1.0 mL时,其线性相关系数为0.994 1,进样量在0.05~0.8 mL时,其线性相关系数为0.994 7,进样量在0.05~0.5 mL时,线性相关系数为0.999 4。因此,进样量在0.05~0.5 mL时有较好的线性,以下试验进样量按0.2 mL进行,试验结果见表1。
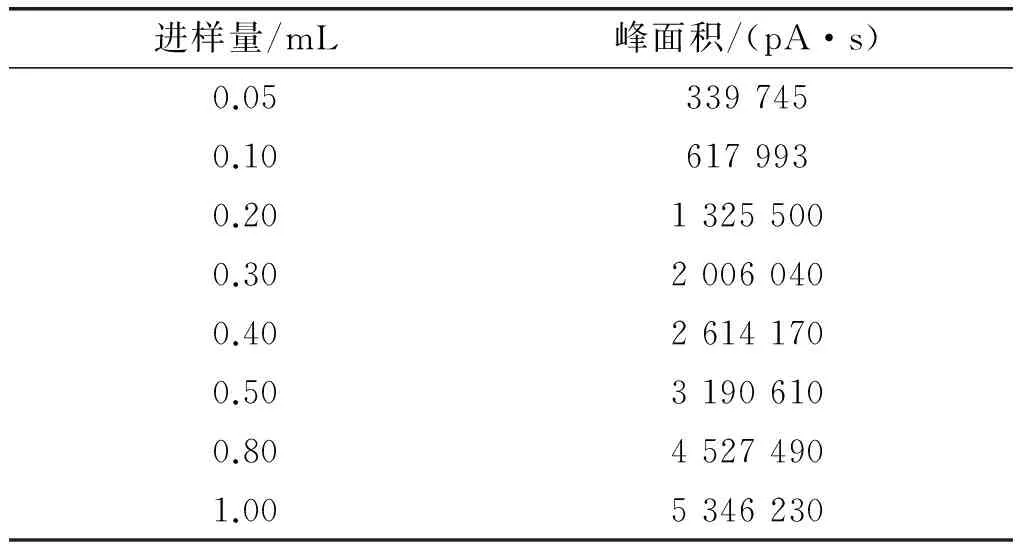
表1 不同进样量与峰面积的对应关系
2.2.4 优化后的气相色谱操作条件
通过操作条件的优化,确定了验证试验的气相色谱操作条件:进样口温度170 ℃、检测室温度250 ℃、柱流量2 mL/min、分流比5 ∶1,柱温初始温度80 ℃保持10 min,以10 ℃/min从80 ℃升温至150 ℃,保持5 min,进样量0.2 mL,以下试验均按此条件进行。
2.3 各有机杂质检出限的测定
采用优化后的气相色谱操作条件进行试验。在已抽过真空的配制瓶中加入含量为10×10-6的各杂质标准品,用干净空气作平衡气,混匀后制成标准样品,进行检出限的测定。1,1-二氟-2-氯乙烯的检出限采用国家药品标准中给出的相对校正因子计算出混合样中的含量,采用空气逐级稀释法测得其检出限。各杂质组分的信噪比见表2,试验结果见图6。
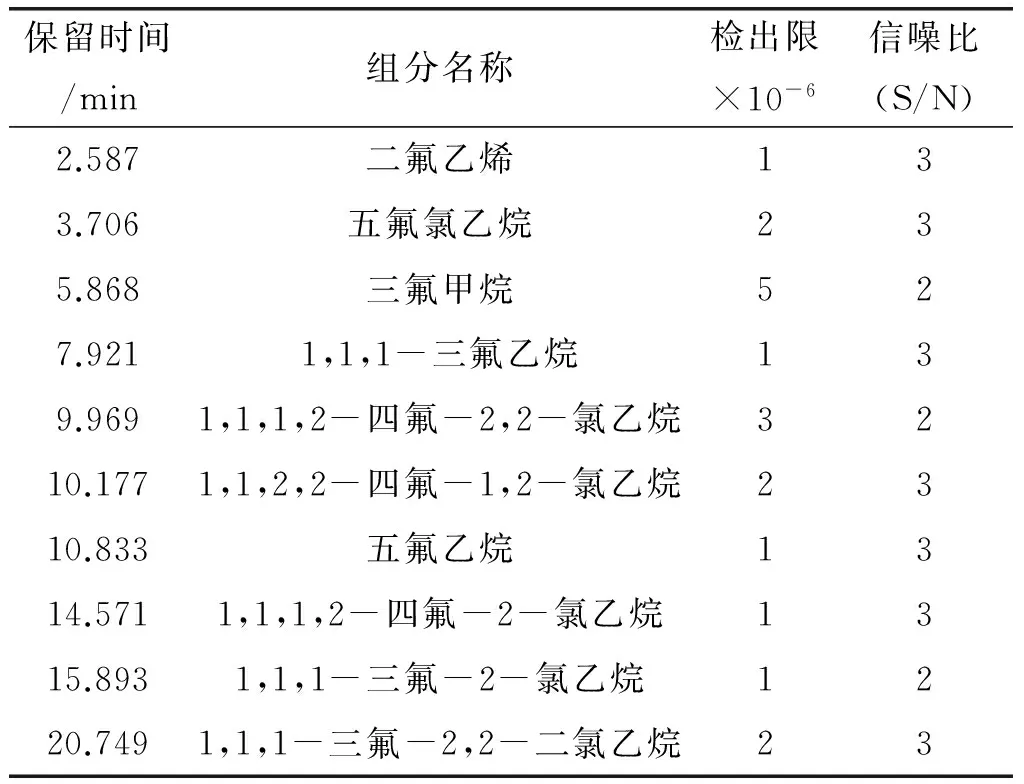
表2 各杂质组分的信噪比测定值
表2显示,各杂质组分的S/N≥2,各杂质组分的检出限符合国家药品标准的要求。
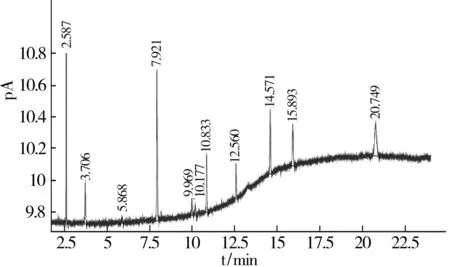
图6 有机杂质检出限谱图(除1,1-二氟-2-氯乙烯)
2.4 主含量及有关杂质的线性试验
2.4.1 药用辅料四氟乙烷的线性试验
在分流条件下,毛细管色谱柱的进样量一般为 0.1~1 mL,通常为0.5 mL以下,本次试验选择0.05 mL、0.1 mL、0.2 mL、0.3 mL、0.4 mL、0.5 mL、0.8 mL、1.0 mL 8个不同的进样量,按优化后的操作条件,分别对同一样品进行试验,结果表明:进样量在0.05~0.5 mL时有较好的线性,其线性相关系数为0.999 4。试验结果见图7。

图7 主含量不同进样量的线性关系图
2.4.2 有关杂质的线性试验
配制10×10-6、20×10-6、30×10-6、40×10-6、50×10-65个分别含二氟乙烯、五氟氯乙烷、三氟甲烷、1,1,1-三氟乙烷、1,1,2,2-四氟-1,2-氯乙烷和1,1,1,2-四氟-2,2-氯乙烷、五氟乙烷、1,1,1,2-四氟-2-氯乙烷、1,1,1-三氟-2-氯乙烷、1,1,1-三氟-2,2-二氯乙烷各有关杂质的标准样品,按优化后的操作条件分别进行测定,各杂质的线性相关系数见表3,结果表明各杂质具有很好的线性关系,线性相关系数均在0.997 0以上。
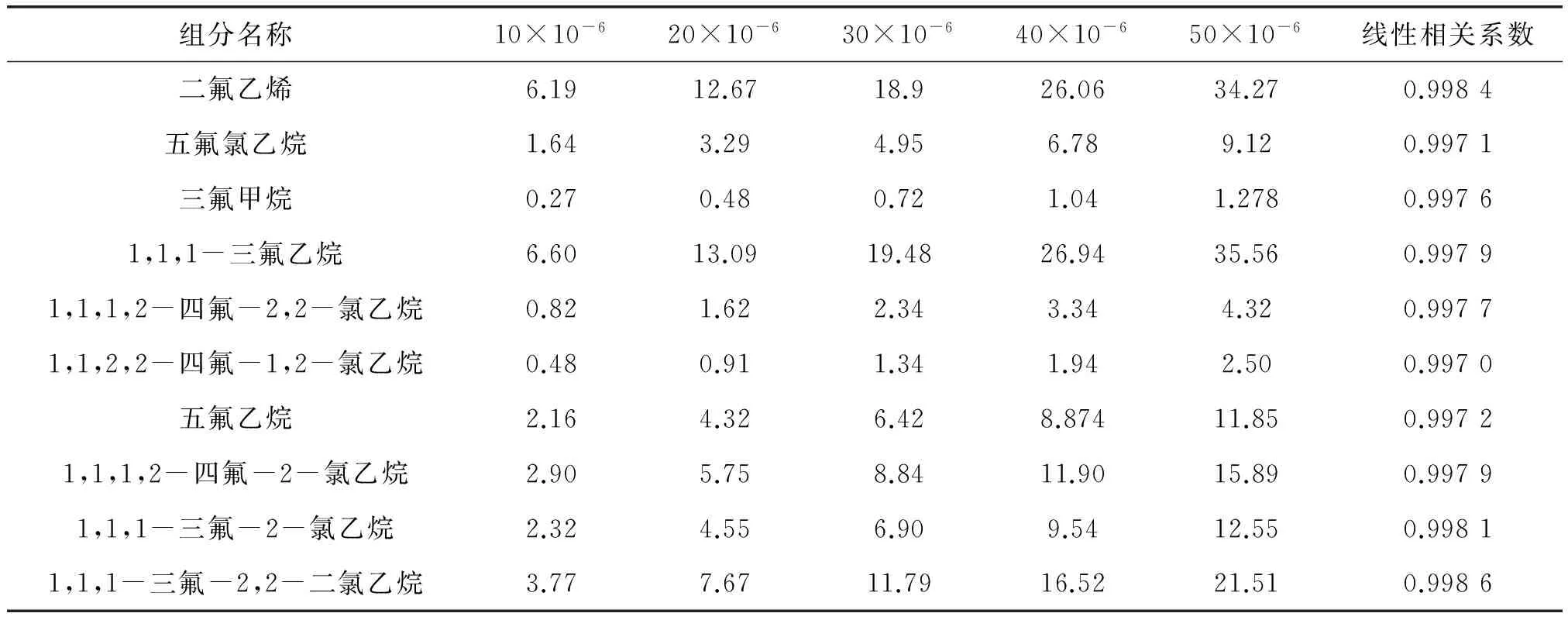
表3 各有关杂质的线性相关系数
2.5 各有机杂质的回收率试验
配制杂质含量分别为0.002 0%和0.003 0%的两个标准样品,按优化后的色谱操作条件,注入色谱仪中进行测定,根据各有机组分含量及峰面积计算标准样品0.002 0%中各组分的相对校正因子,以标准样品0.0030%为加标样进行回收率的计算。计算结果见表4,其回收率为96.3%~102.5%,结果表明本方法准确性高。
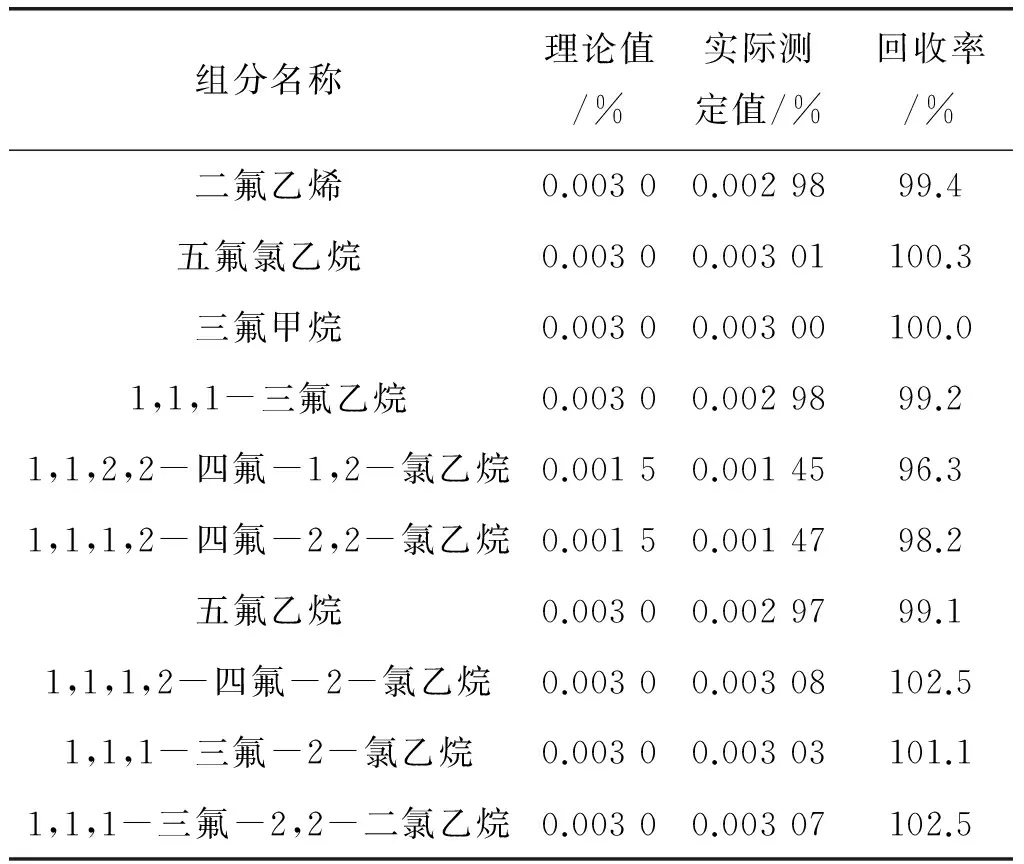
表4 各杂质组分的回收率试验
2.6 精密度试验
采用连续3个批次的样品分别进行8平行试验,试验结果见表5。
从表5可以看出:该方法重复性稳定,标准偏差小,能满足分析的需要。

表5 1,1,1,2-四氟乙烷含量的精密度试验(质量分数/%)
3 结论
通过验证试验,发现国家药品标准在关于含量及有关物质的色谱操作条件选择不是很合适,我们按试验结果重新确定了主含量及有关杂质的最佳操作条件。采用优化后的国家药品标准测定主含量和有关物质,分离度更高,检出限低(除三氟甲烷外,各杂质均小于3×10-6),主含量的线性相关系数为0.999 4,有关杂质的线性相关系数均大于0.997 0,回收率在96.3%~102.5%之间,精密度标准偏差小。该方法能满足药用辅料检测的需要。
[1] WS5-FG-003-2013, 国家药品标准四氟乙烷[S]. 国家食品药品监督管理总局, 2013.
Research on the Method of Pharmaceutical HFA-134a of National Standard
Chen Caiqin, Liu Hongxiu, Dai Meng
(Zhejiang Quhua Fluoro-Chemistry Co., Ltd., Quzhou 324004, China)
To study the pharmaceutical HFA-134a standards for purity analysis method, analyzed the shortcomings of operating conditions, to optimize the operation conditions of chromatography, investigated the method of detection limit, linear recovery rate and precision. The results show that the optimized method has better resolution and higher accuracy.
pharmaceutical HFA-134a; national standard; chromatographic operating conditions; sample injection
陈彩琴(1977—),女,工程师,从事氟制冷剂系列产品质量检验管理工作。
猜你喜欢
分析仪器(2022年5期)2022-10-14
浙江化工(2022年8期)2022-09-05
中国石油大学学报(自然科学版)(2022年4期)2022-09-05
煤气与热力(2021年3期)2021-06-09
化工管理(2021年7期)2021-05-13
中国船检(2019年6期)2019-07-24
铜仁学院学报(2018年6期)2018-07-05
浙江化工(2015年4期)2015-11-28
浙江化工(2012年2期)2012-01-11
Defence Technology(2010年3期)2010-03-09
