
KCNQ4和GJB2基因多态性与职业噪声性听力损失的相关性研究
2015-02-08 02:01谈柯宏于德财张娟尹立红浦跃朴
癌变·畸变·突变 2015年4期
谈柯宏,于德财,张娟,*尹立红,浦跃朴
(东南大学公共卫生学院环境医学工程教育部重点实验室,江苏南京210009)
职业性噪声导致的听力损失是世界范围内普遍存在的职业损害之一,是由于长期高水平接触噪声而发生的一种进行性感音性听力损伤。职业噪声性听力损失占全球范围内成人听力损失的16%,仅次于老年性耳聋,其中发展中国家明显高于发达国家[1]。人群流行病学研究表明,暴露于相同噪声环境下,工人出现噪声性听力损失存在个体差异,噪声性听力损失(noise-induced hearing loss,NIHL)的发生是噪声暴露与遗传因素共同作用的结果,其中遗传性因素对于发生听力损失的作用超过50%[2]。目前噪声性听力损失机制假说较多,主要包括局部缺血、神经因子缺乏、钾钙离子平衡失调、过量自由基的产生、螺旋器细胞间连接功能的中断以及缝隙连接蛋白的结构改变等[3]。由于内耳离子稳态平衡对于维持听觉和平衡具有重要影响,因而与离子平衡相关的基因研究较为透彻[4]。内耳淋巴循环含有较高浓度K+,参与电信号的传导,如果K+通道及相关协同转运蛋白发生改变,可以导致内耳钾离子转运机制紊乱进而出现听力损伤[5]。故本研究选取钾离子通道蛋白基因KCNQ4 rs34287852和缝隙连接蛋白基因GJB2 rs3751385,探讨其基因多态性与中国汉族人群噪声性听力损失的相关性。
1 对象与方法
1.1 对象
1.1.1 病例选择南京市某汽车制造厂及某轨道交通车辆厂发生噪声性听力损伤的工人103例。纳入标准:依据《GBZ 49-2007 职业性听力损失诊断标准》对职业性听力损失进行诊断,双耳高频(3000、4000、6000 Hz)平均听阈≥40 dB(A),且双耳听力曲线符合NIHL特点。同时符合下列条件:无耳毒性化学物接触史,无爆震史、家族性耳聋史及耳毒性药物使用史。
1.1.2 对照选择与病例组工人相同作业岗位,双耳高频、语频平均听阈≤25 dB(A)的工人,按同性别、同年龄(±5岁),同接噪工龄(±3年)的要求1∶1配对。
1.1.3 试剂和仪器基因组DNA提取试剂盒(DP319),购自天根生化科技(北京)有限公司;引物设计与合成,上海捷瑞生物工程有限公司。凝胶成像分析系统(Multiphor II型);基因学分析仪(ABI 3730)。
1.2 基因组DNA提取
对符合要求的研究对象,抽取其外周血2 mL。使用血液基因组提取试剂盒提取基因组DNA,紫外分光光度计测量DNA浓度和纯度,经鉴定合格,进行下一步实验。
1.3 基因分型
1.3.1 引物序列KCNQ4基因:上游引物5´-CCCA AGTCCTAAGTCAGCTTTGTC-3´,下游引物5´-GAG TCTCAGAGATGCCCGGAAG-3´,扩增产物287 bp。GJB2基因:上游引物5´-AGAAATAGACAGCATGAG AGGGATG-3´,下游引物5´-CTTTGTTTTGAGGCTT TAGGGGA-3´,扩增产物180 bp。
1.3.2 PCR扩增扩增体系:使用PCR kit(Takara Ex Taq),总体积25 μL,其中TaqDNA聚合酶1 U(5 U/μL),Buffer缓冲液(含Mg2+)4 μL,脱氧核苷三磷酸(dNTPs) 1.6 μL (2.5 mmol/L),引物各0.4 μL (10 μmol/L),模板50 ng。反应条件:94 ℃预变性5 min,94 ℃变性1 min、60 ℃退火30 s、72 ℃延伸30 s,35个循环,最后72 ℃延伸10 min。
1.3.3 基因型鉴定采用琼脂糖凝胶电泳,经Multiphor II型凝胶成像分析系统确认扩增目的片段。对扩增产物进行基因分析(华大基因),鉴定目的位点基因型。
1.4 统计学分析
使用SPSS 19.0统计软件,对两位点各基因型和等位基因在两组人群中的分布频率进行χ2分析,使用Logistic回归分析,计算突变基因型根据两组突变比值比(odds ratio,OR)比较突变基因型噪声性听力损失发生的危险度,检验水准α=0.05。
2 结果
2.1 病例组与对照组一般人口学特征
例组与对照组一般信息如表1所示,两组工人年龄和接噪工龄差异均无统计学意义(P均>0.05)。
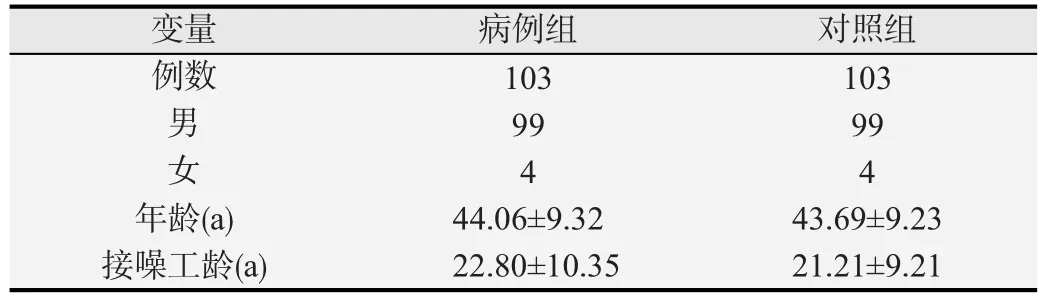
表1 病例组与对照组年龄、性别、工龄、接噪工龄构成情况
2.2 KCNQ4和GJB2基因型测序
基因型测序结果见图1,KCNQ4 rs34287852经正向测序,基因型包含野生纯合型TT(A1)、突变杂合型GT(A2)、突变纯合型GG(A3)。GJB2 rs3751385经反向测序,其基因型包含野生纯合TT(B1)、突变杂合型CT(B2)、突变纯合型CC(B3)。
2.3 KCNQ4和GJB2基因型与噪声性听力损失
KCNQ4 rs34287852和GJB2 rs3751385基因型及等位基因频率在病例和对照组间分布见表2。KCNQ4 rs34287852位点T、G等位基因在病例组中分布频率分别为93.2%和6.8%,在对照组中分别为95.6%和4.4%,其中TT、GT、GG基因型在病例组和对照组的分布频率分别为 88.3%、9.7%、1.9%和91.3%、8.7%、0。KCNQ4突变位点(rs34287852)基因型频率和等位基因频率在两组之间的分布差别均无统计学意义(P=0.479,P=0.394)。GJB2 rs3751385位点T、C等位基因在病例组中分布频率分别为46.6%和53.4%,在对照组中分别为57.8%和42.2%,等位基因频率在两组之间分布差异具有统计学意义(P=0.03);TT、CT、CC 3种基因型在病例组和对照组的分布频率分别为17.4%、58.3%、24.3%和31.1%、53.4%、15.5%;病例组GJB2 rs3751385突变基因型频率更高,差异有统计学意义(P=0.047)。工人GJB2 rs3751385突变纯合型CC相比野生纯合型TT发生噪声性听力损失的危险性大(OR=2.78,95% CI=1.18~6.52)。
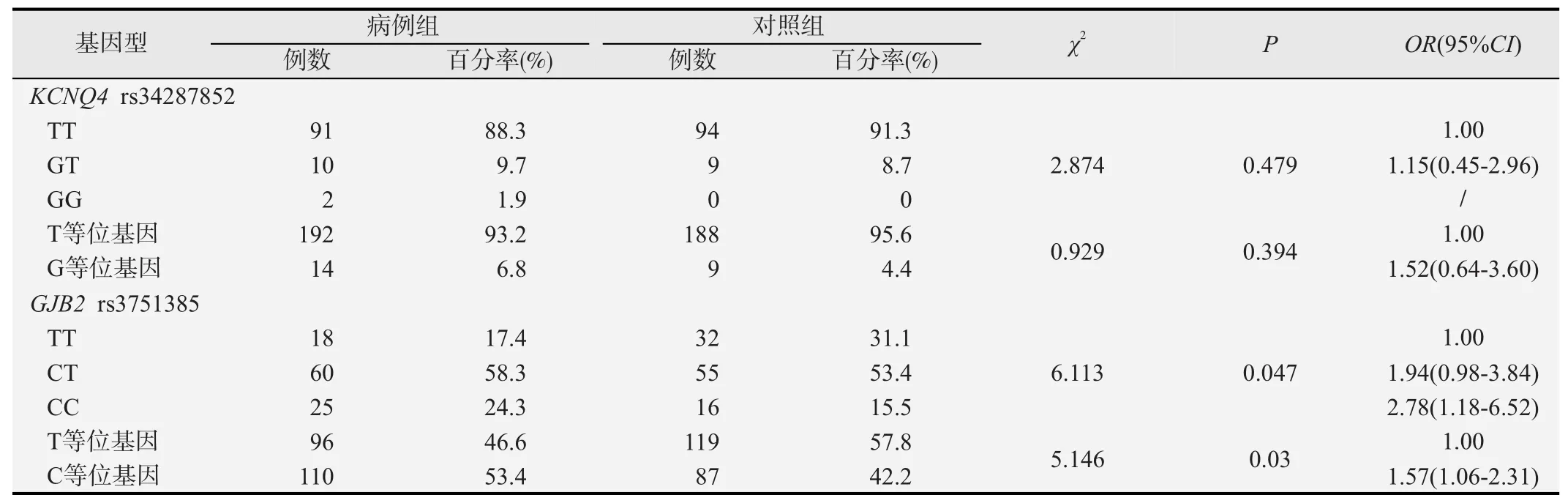
表2 KCNQ4和GJB2基因多态性与噪声性听力损失的相关分析
3 讨论
参与调控内耳淋巴液离子转运通道的蛋白主要包含KCNQ1,KCNQ4等K+通道蛋白,构成电压门控式通道,控制内耳毛细胞内外K+平衡。其中KCNQ4基因参与耳蜗外毛细胞和前庭器官毛细胞功能,其通道存在于内耳外毛细胞膜外侧,控制K+外流,维持细胞静息电位[6-7]。其突变可导致常染色体显性慢性进行性听力损伤,从高频听阈发展覆盖各频段听阈[8-9]。因而其突变是否对于噪声性听力损伤的发生有贡献,对于筛查噪声作业易感基因具有重要意义。在瑞典和波兰两例噪声作业工人病例对照研究中,KCNQ4 rs34287852位点突变与噪声作业存在统计学差异[10-11]。但其最小等位基因频率(minor allele frequency,MAF)为22.2%,说明位点突变在白种人群广泛存在。在本研究中病例组和对照组中MAF分别为6.8%和4.4%,该位点突变可能存在种群差异。研究结果表明KCNQ4 rs34287852在两组人群中突变基因型及等位基因频率差异均无统计学意义,其突变不影响噪声性听力损失的发生。
内耳细胞间存在多聚体结构的缝隙连接蛋白Connexins(Cx),其对于维持内耳淋巴液成分稳定,特别是在K+传递中具有重要作用[12-13]。其家族蛋白基因包括GJB2和GJB6基因突变确认与人听力损失相关[14-15]。其中GJB2基因约有220个突变位点与听力损伤存在相关,50%常染色体隐性非综合性听力损失(autosomal recessive non-syndromic hearing loss,ARNSHL)与其相关,且多为迟发型听力损伤[16]。在对GJB2基因突变与噪声性听力损失的研究中,2006年瑞典噪声暴露工人病例对照研究认为选取GJB2基因两个突变位点rs3751385,rs5030700的基因型与噪声性听力损失无相关性[10],但在2009年波兰噪声作业工人配对病例研究中,发现rs3751385基因型与噪声性听力损失存在相关性[11],这可能与该位点在两人群中等位基因频率不同及连锁不平衡影响[17]。这一位点突变与ARNSHL的相关性在希腊人和葡萄牙人得到验证[18-19]。本研究中103例噪声性听力损失工人GJB2 rs3751385 突变的等位基因和基因型频率的差异有统计学意义,其杂合突变在噪声作业人群中的噪声性听力损失风险为野生型2.78倍,突变与噪声性听力损失存在相关性。现在已知基因的3´UTR区域对于转录物断裂、选择性多腺苷酸化以及mRNA的出核起着重要调控作用[20]。rs3751385位于GJB2基因的3´UTR,有研究表明该突变可能通过调控GJB2基因的表达来影响听力[21]。
综上,KCNQ4 rs34287852位点突变较少,其突变与噪声性听力损失无相关性,这也可能受限于样本量的比例。GJB2 rs3751385位点突变与噪声性听力损失可能存在相关性,可以进一步从机制上去验证这一位点突变与噪声性听力损失的关系,从而为噪声性听力损失的预防提供科学依据。
[1] Nelson DI,Nelson RY,Concha-Barrientos M,et al. The global burden of occupational noise-induced hearing loss[J]. Am J Ind Med,2005,48(6):446-458.
[2] Sliwinska-Kowalska M,Davis A. Noise-induced hearing loss[J]. Noise & Health,2012,14(61):274-180.
[3] 曲腾飞,龚树生. 噪声性聋致病机制及其防护研究[J]. 国际耳鼻咽喉头颈外科杂志,2013,37(006):318-320.
[4] 王秋菊,Mohamed A. 耳内科疾病相关基础研究与诊治新进展:上篇[J]. 中 华耳科学杂志,2012,10(2):201-207.
[5] Wangemann P. K+cycling and the endocochlear potential[J].Hear Res,2002,165:1-9.
[6] Kubisch C,Schroeder BC,Friedrich T,et al. KCNQ4,a novel potassium channel expressed in sensory outer hair cells,is mutated in dominant deafness[J]. Cell,1999,96(3):437-446.
[7] Kharkovets T,Harderlin JP,Safieddine S,et al. KCNQ4,a K+channel mutated in a form of dominant deafness,is expressed in the inner ear and the central auditory pathway[J].Proc Natl Acad Sci USA,2000,97(8):4333-4338.
[8] Naito T,Nishio SY,Iwasa Y,et al. Comprehensive genetic screening of KCNQ4 in a large autosomal dominant nonsyndromic hearing loss cohort:genotype-phenotype correlations and a founder mutation[J]. PLoS One,2013,8(5):e63231.
[9] Nie L. KCNQ4 mutations associated with nonsyndromic progressive sensorineural hearing loss[J]. Curr Opin Otolaryngol Head Neck Surg,2008,16(5):441-444.
[10] Van Laer L,Carlsson PI,Ottschytsch N,et al. The contribution of genes involved in potassium-recycling in the inner ear to noise-induced hearing loss[J]. Hum Muta,2006,27:786-795.
[11] Pawelczyk M,Van Laer L,Fransen E,et al. Analysis of gene polymorphisms associated with K on circulation in the inner ear of patients susceptible and resistant to noise-induced hearing loss[J]. Ann Hum Genet,2009,73(4):411-421.
[12] Kikuchi T,Adams JC,Miyabe Y,et al. Potassium ion recycling pathway via gap junction systems in the mammalian cochlea and its interruption in hereditary nonsyndromic deafness[J]. Med Electron Microsc,2000,33(2),51-56.
[13] Zhao,HB,Kikuchi T,Ngezahayo A,et al. Gap junctions and cochlear Homeostasis[J]. J Mem Biol,2006,209(2/3),177-186.
[14] Richard G,White TW,Smith LE,et al. Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma[J].Hum Genet,1998,103(4):393-399.
[15] Grifa A,Wagner CA,D’Ambrosio L,et al. Mutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locus[J]. N at Genet,1999,23(1):16-18.
[16] Zelante L,Gasparini P,Estivill X,et al. Connexin26 mutations associated with the most common form of nonsyndromic neurosensory autosomal recessive deafness(DFNB1) in Mediterraneans[J]. Hum Mol Genet,1997,6(9):1605-1609.
[17] Sliwinska-Kowalska M,Pawelczyk M. Contribution of genetic factors to noise-induced hearing loss:a human studies review[J]. Mutat Res,2013,752(1):61-65.
[18] Kokotas H,Van Laer L,Grigoriadou M,et al. Strong linkage disequilibrium for the frequent GJB235delG mutation in the Greek population[J]. Am J Med Genet,2008,146(22):2879-2884.
[19] Grillo AP,de Oliveira FM,de Carvalho GQ,et al. Single nucleotide polymorphisms of the GJB2 and GJB6 genes are associated with autosomal recessive nonsyndromic hearing loss[J]. Biomed Res Int,2015. d oi:10.1155/2015/318727.
[20] Grzybowska EA,Wilczynska A,Siedlecki JA. Regulatory functions of 3´-UTRs[J]. Biochem Bioph Res Co,2001,288(2):291-295.
[21] Matos TD,Simões-Teixeira H,Caria H,et al. Assessing noncoding sequence variants of GJB2 for hearing loss association[J]. Genet Res Int,2011. d oi:10.4061/2011/827469.
猜你喜欢
传染病信息(2022年4期)2022-11-23
分子催化(2022年1期)2022-11-02
西部医学(2022年9期)2022-09-26
智慧健康(2021年17期)2021-07-30
烟草科技(2021年6期)2021-06-24
生物学教学(2018年4期)2018-11-29
新课程·下旬(2018年9期)2018-11-14
电脑知识与技术(2018年19期)2018-11-01
生物学教学(2018年8期)2018-09-03
青少年科技博览(中学版)(2015年10期)2015-01-11
