
高效液相色谱法同时测定阿霉素/姜黄素载双药纳米粒的药物含量及包封率
2015-01-19 02:28:42林红霞戴东波丁玎王茜
浙江医学 2015年10期
林红霞 戴东波 丁玎 王茜
高效液相色谱法同时测定阿霉素/姜黄素载双药纳米粒的药物含量及包封率
林红霞 戴东波 丁玎 王茜
目的 建立采用高效液相色谱法(HPLC)同时测定阿霉素/姜黄素载双药纳米粒(DOX/CUR-NPs)中阿霉素和姜黄素的含量及包封率的方法。方法 采用乳化溶剂挥发法制备DOX/CUR-NPs;按照制剂含量测定方法学要求考察所建立HPLC法的专属性、精密度、回收性等,并结合超速离心法测定DOX/CUR-NPs的包封率。结果 成功制备了DOX/CUR-NPs;在建立的色谱条件下,DOX和CUR专属性好,精密度、回收率、重复性等试验均符合方法学要求。两药在0.5~50.0μg/ml浓度范围内线性关系良好(r=0.9999),纳米粒中DOX与CUR的包封率分别为(58.9±1.11)%和(75.4±1.76)%。结论 HPLC法准确可靠、简单快速,可用于DOX/CUR-NPs的药物含量及包封率的同时测定。
阿霉素 姜黄素 载双药纳米粒 含量测定 高效液相色谱
盐酸阿霉素(DOX)属水溶性蒽环类抗肿瘤抗生素,其抗癌谱广、活性强、疗效确切,但其耐药现象(MDR)普遍[1],限制了其在临床上的应用。姜黄素(CUR)是姜黄属中药的主要药理成分,能抑制癌细胞的转移、增殖,下调P-糖蛋白、多药耐药相关蛋白1和乳腺癌耐药相关蛋白,以多途径逆转肿瘤多药耐药,是一种极具开发前景的MDR中药逆转剂[2-3]。笔者在研制盐酸阿霉素/姜黄素载双药纳米粒(DOX/CUR-NPs)的过程中,发现现有的研究多为分开测定两药的含量,操作繁琐,耗时耗力[4-5]。因此,本研究建立了一种同时测定纳米粒中DOX、CUR药物含量的高效液相法,用于制剂优化过程中包封率、载药量的定量测定,现报道如下。
1 材料和方法
1.1 材料 DOX为浙江海正制药有限公司赠予;姜黄素购自成都普思生物技术有限公司;DOX、CUR标准品均购自中国药品生物制品检定所;mPEG-PLA购自山东岱罡生物科技有限公司;PVA购自百灵威科技有限公司;乙腈、甲醇均购自美国Honeywell Burdick&Jackson公司。Agilent1200高效液相色谱仪购自美国Agilent公司;OptimaMAX超速离心机购自美国Beckman公司;99-ⅡDN型超声波细胞粉碎机购自宁波新芝生物科技股份有限公司;CP225D电子天平购自德国赛多利斯公司。
1.2 方法
1.2.1 DOX/CUR-NPs及空白NPs的制备 精密称取10 mg的DOX溶于1ml双蒸水,混匀,吸取100μl构成内水相;精密称取5mg的CUR和50mg的mPEG-PLA,溶于2ml二氯甲烷-丙酮(3∶2)混合溶剂中,构成有机相;内水相与有机相混合后,冰浴中间歇式超声(300W,60s)得到初乳;另量取4ml 3%的PVA水溶液,构成外水相;初乳与外水相混合后,冰浴中间歇式超声(400W,60s)得到复乳;将此复乳缓慢滴加至高速磁力搅拌下的20ml 0.5%PVA水溶液中,滴毕,继续搅拌10min,室温减压蒸去有机溶剂和部分水,得到带桔黄色乳光的纳米粒混悬液(DOX/CUR-NPs)。按上述方法,仅不加DOX与CUR,制备空白NPs混悬液。
1.2.2 色谱条件 色谱柱:ZORBAX Eclipse XDB C18柱(250mm×4.6mm,5 μm);流动相:以乙腈为A相,以0.02 mol/L磷酸二氢钠(磷酸调节pH至3.0)为B相,梯度洗脱:0~6min,30%A;6~7min,30%~60%A;7~15 min,60%A;体积流量1.0ml/min;柱温25℃;进样量20 μl,检测波长:0~7min,233nm;7~15min,425nm。
1.2.3 溶液的制备 分别精密称取DOX对照品、CUR对照品各10mg置于50ml棕色量瓶中,甲醇溶解,并稀释至刻度,制成DOX、CUR质量浓度各为200μg/ml的对照品储备液。分别精密量取DOX、CUR对照品储备液各25ml置50ml棕色量瓶中,混匀,即为DOX、CUR质量浓度各为100μg/ml的混合对照品储备液。精密量取制备的DOX/CUR-NPs混悬液1ml于10ml棕色量瓶中,加甲醇稀释至刻度,超声,0.22μm微孔滤膜过滤,续滤液即为供试品溶液。精密量取制备的空白纳米粒混悬液,按上述供试品溶液的制备法操作,即得空白纳米粒对照液。
1.3 专属性考察 分别量取20μl“1.2.3”项下DOX对照品储备液、CUR对照品储备液、混合对照品储备液、空白纳米粒对照液和供试品溶液,按“1.2.2”项下色谱条件进样测定,考察仪器专属性。
1.4 标准曲线的绘制 分别精密量取20μl混合对照品储备液置于10ml棕色量瓶中,甲醇稀释至刻度,得质量浓度分别为0.5、1.0、2.5、5、10、25、50 μg/ml的系列混合对照品溶液,按“1.2.2”项下色谱条件进样检测,以峰面积A对质量浓度C进行线性回归,建立标准曲线方程。
1.5 精密度试验 取低(0.5μg/ml)、中(5μg/ml)、高(50μg/ml)3个浓度的混合对照品溶液,按“1.2.2”项下色谱条件进样测定,分别于1d内测定5次,连续测定5d,计算得日内和日间精密度。
1.6 回收率试验 吸取同一批次制备的空白纳米粒混悬液1ml于10ml棕色量瓶中,分别精密加入混合对照品溶液适量,用甲醇稀释至刻度,配成质量浓度分别为低(0.5μg/ml)、中(5μg/ml)、高(50μg/ml)3个浓度的样品溶液(各浓度样品平行制备3份)。经0.22μm微孔滤膜滤过,取续滤液按“1.2.2”项下色谱条件进样测定,每个浓度平行操作3次,计算得DOX、CUR的回收率。
1.7 重复性试验 取同一批次制备的DOX/CUR-NPs混悬液平行制备6份供试品溶液,按“1.2.2”项下色谱条件进样测定,计算相对标准偏差(RSD)。
1.8 稳定性试验 取同一批次制备的DOX/CUR-NPs供试品溶液,分别于0、2、4、8、6、12、24h按“1.2.2”项下色谱条件进样测定,计算RSD。
1.9 超速离心回收率试验 精密量取混合对照品储备液1ml至10ml棕色量瓶中,空白纳米粒混悬液稀释至刻度,配制成含DOX、CUR均为0.5、5、50μg/ml的溶液,各3份。精密量取各溶液4ml置于离心管中,封口,超速离心(4℃,40 000r/min,60min)后,取上清液经0.22 μm微孔滤膜过滤,取续滤液按“1.2.2”项下色谱条件进样测定,计算超速离心回收率。
1.10 纳米粒中药物的含量测定 精密量取 DOX/ CUR-NPs混悬液1ml于10ml棕色量瓶中,加甲醇稀释至刻度,超声,0.22μm微孔滤膜过滤,取续滤液按“1.2.2”项下色谱条件进样测定,重复3次,计算得药物含量。
1.11 包封率的测定 精密量取DOX/CUR-NPs混悬液4ml于具塞离心管中,超速离心(4℃,40 000r/min,60min)后,取上清液1ml,置10ml量瓶中,用甲醇稀释至刻度,经0.22μm微孔滤膜滤过,取续滤液按“1.2.2”项下色谱条件进样测定,计算游离DOX、CUR含量,分别记作W1、W2;另精密量取DOX/CUR-NPs混悬液1ml,置10ml量瓶中,用甲醇稀释至刻度,超声,经0.22 μm微孔滤膜滤过,取续滤液按“1.2.2”项下色谱条件进样测定,计算纳米粒混悬液中总的药物含量,记作W3、W4。按下式计算纳米粒的包封率:
包封率(DOX)%=(W3-W1)/W3×100%包封率(CUR)%=(W4-W2)/W4×100%
2 结果
2.1 专属性考察 DOX对照品储备液、CUR对照品储备液、混合对照品储备液、空白纳米粒对照液和供试品溶液的高效液相色谱图见图1。
由图1可知,DOX、CUR色谱峰峰型良好,辅料和溶剂对药物测定无干扰。
2.2 标准曲线 试验重复6次,计算得DOX标准曲线方程:A=83.994C-8.8053,CUR标准曲线方程:A= 191.07C+56.769,表明DOX、CUR浓度在0.5~50 μg/ml范围内与峰面积呈良好的线性关系,详见图2。
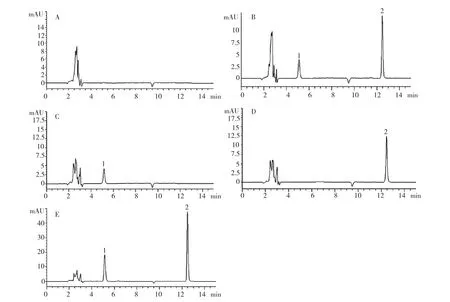
图1 高效液相色谱图(A:空白NPs;B:DOX/CUR对照品;C:DOX对照品;D:CUR对照品;E:DOX/CUR-NPs;1:DOX,2:CUR)

图2 DOX、CUR标准曲线图
2.3 精密度试验 低、中、高3个浓度的混合对照品溶液中,DOX的日内精密度RSD分别为1.14%、1.05%、1.02%,日间精密度RSD分别为1.43%、1.20%、0.63%;CUR的日内精密度RSD分别为1.36%、0.80%、0.55%,日间精密度RSD分别为1.64%、1.47%、0.64%,均符合方法学要求(RSD≤2%)。
2.4 回收率试验 低、中、高3个浓度的混合对照品溶液中,DOX的回收率分别为96.7%、102.3%、98.0%,RSD分别为1.57%、1.10%、1.76%;CUR的回收率分别为104.3%、95.7%、102.5%,RSD分别为1.31%、1.47%、1.35%,均符合方法学要求。
2.5 重复性、稳定性试验 重复进样6次,结果DOX与CUR含量的RSD分别为1.30%和1.14%,符合方法学要求。在24h内,测得DOX的RSD为1.15%,CUR的RSD为1.03%,表明样品稳定性良好,均符合方法学要求。
2.6 超速离心回收率 超速离心法可实现纳米粒与游离药物的分离,低、中、高3个浓度的超速离心回收率见表1。
表1 游离药物与纳米粒的分离结果
2.7 药物含量和包封率测定 DOX/CUR-NPs中DOX含量为45.5μg/ml,RSD为1.28%;CUR含量为233.2μg/ ml,RSD为 1.42%;DOX与 CUR的包封率分别为(58.9±1.11)%和(75.4±1.76)%,详见表2。

表2 含量和包封率测定结果
3 讨论
多药耐药是导致肿瘤化疗失败的主要原因之一[1]。纳米粒系一种新型药物载体,有学者尝试将DOX与CUR同时包裹于纳米粒,发现可在较大程度上逆转MDR,同时降低了其对正常细胞的毒性[4-5],故研制DOX/CUR-NPs具有较大的意义。但DOX与CUR的水溶性相差较大。Jinghua等[4]分别采用HPLC法来测定纳米粒中DOX与CUR的含量,而Ranjita等[5]则采用荧光分光光度计法和HPLC法分别测定两药含量,均操作繁琐,耗时耗力。本文通过多次筛选,最终确定以乙腈-0.02mol/L磷酸二氢钠(磷酸调节pH至3.0)作为流动相,梯度洗脱,检测波长在相应时间调整为DOX最大吸收波长233nm及CUR最大吸收波长425 nm。在此色谱条件下,DOX与CUR分离良好,保留时间合适,辅料及溶剂不干扰测定,结果准确,回收率高,重复性好,为DOX/CUR载双药系统中药物含量测定提供了一种简便、快速、有效的方法。
包封率的测定方法主要有超速离心法,透析法,超滤离心法和凝胶色谱法等[6-8]。后两者大多用于脂质体包封率的测定,而透析法透析过程缓慢,易造成药物的泄露且比较费时;故本实验采用低温超速离心法分离药物与纳米粒。回收率实验表明,该离心条件满足实验要求,达到了较好的分离效果。
本实验仅采用了紫外分光检测器,其测得药物的线性范围为0.5~50 μg/ml,难以达到药物在体内的分析要求。而DOX与CUR均具有荧光特性,故下一步可采用荧光分光检测器,提高灵敏度,为DOX/CUR载双药纳米粒的体内药动学研究奠定基础。
[1] Perez-Tomas,Ricardo.Multidrug resistance:retrospect and prospects in anti-cancer drug treatment[J].Curr Med Chem,2006,13 (16):l859-1876.
[2] Hatcher H,Planalp R,Cho J,et al.Curcumin:from ancient medicine to current clinical trials[J].Cell Mol Life Sci,2008,65(11): 1631-1652.
[3] Um Y,Cho S,Woo H,et al.Synthesis of curcumin mimics with multidrug resistance reversal activities[J].Bioorg Med Chem, 2008,16(7):3608-3615.
[4] Jinghua D,HeidiMM,Zhang YD,et al.Reversion ofmultidrug resistance by co-encapsulation of doxorubicin and curcumin in chitosan/poly(butyl cyanoacrylate)nanoparticles[J].Int J Pharm, 2012,426(1):193-201.
[5] Ranjita M,Sanjeeb K S.Coformulation of doxorubicin and curcumin in poly (D,L-lactide-co-glycolide)nanoparticles suppresses the development of multidrug resistance in K562 cells[J]. MolPharm,2011,8(3):852-866.
[6]戴玉娇,符华林,杨乐,等.RP-HPLC法测定磷酸泰乐菌素脂质体中药物含量及包封率[J].药物分析杂志,2010,30(12):2299-2303.
[7]代文婷,张典瑞,郭晨煜,等.HPLC法测定冬凌草甲素纳米脂质载体药物的含量及包封率[J].药物分析杂志,2009,29(4):587-590.
[8]曾昭武,周广林,展晓日,等.超高效液相色谱法测定榄香烯脂质体的含量及包封率[J].药物分析杂志,2010,30(3):508-510.
Simultaneous determination of content and entrapment efficiency of doxorubixin and curcumin loaded nanoparticles by HPLC
LIN Hongxia, DAI Dongbo,DING ding,et al.
Department of Clinical Laboratory,Wenlin First People's Hospital,Wenling 317500,China
【 Abstract】 Objective To establish a HPLC method for the simultaneous determination of content and entrapment efficiency of doxorubixin and curcumin loaded mPEG-PLA nanoparticles(DOX/CUR-NPs). Methods The DOX/CUR-NPs were prepared by emulsion-solvent evaporation method.The specificity,precision,recovery and other tests of the established HPLC method were investigated according to analysis methodology for pharmaceutical preparations.The entrapment efficiency was determined by HPLC combined with centrifugal ultrafiltration. Results The DOX/CUR-NPs were prepared successfully.Under the HPLC conditions established,specificity,precision,recovery,repeatability of DOC and CUR met the challenge of analysis methodology.A good linear relationship was found between the peak area and the concentration of DOX and CUR ranging from 0.5μg/ml to 50.0μg/ml(r=0.9999),respectively.The entrapment efficiency of DOX and CUR in nanoparticles was (58.9±1.11)%and (75.4±1.76)%,respectively. Conclusion The established method is simple,accurate,sensitive and suitable for simultaneous determination of content and entrapment efficiency of doxorubixin and curcumin loaded nanoparticles.
Doxorubixin Curcumin Dual-loaded nanoparticles Content determination HPLC
2014-12-29)
(本文编辑:严玮雯)
317500 温岭市第一人民医院检验科(林红霞、丁玎),药剂科(戴东波、王茜)
戴东波,E-mail:linhx38_5@yeah.net
猜你喜欢
宁夏医科大学学报(2022年5期)2022-11-30 23:32:05
宁夏医科大学学报(2022年6期)2022-11-22 08:33:29
宁夏医科大学学报(2022年8期)2022-11-22 03:13:47
Medical Data Mining(2019年2期)2019-07-16 04:36:08
山东化工(2018年15期)2018-09-20 08:55:34
中成药(2018年2期)2018-05-09 07:19:43
中成药(2018年3期)2018-05-07 13:34:37
临床肝胆病杂志(2017年1期)2017-03-07 03:04:26
首都食品与医药(2015年18期)2015-11-03 05:59:08
西南军医(2015年1期)2015-01-22 09:08:25
