
关于可变剪接的相关论述
2015-01-13 10:17:34万丽娜
宜春学院学报 2015年12期
万丽娜,马 猛
(安徽大学 计算机科学与技术学院,合肥 230601)
1 可变剪接的研究
mRNA 前体通过不同的剪接方式(即不同剪切位点和拼接方式)产生不同的mRNA 异构体(isoform),出现一个基因指导合成多种不同的蛋白质产物。基因可变剪接的主要模式有[1](1)盒式外显子(2)内含子保留(3)互斥外显子(4)5’供体位点可变(5)3’受体位点可变(6)可变起始外显子(7)可变终止外显子。如图1 所示。

图1 可变剪接模式
对可变剪接的研究也在不断的发展中,其中主要的方法有基于比较基因组学的可变剪接研究、基于高通量测序的可变剪接研究和基于基因组序列特征的可变剪接研究。
1.1 基于比较基因组学的可变剪接研究
比较基因组学(Comparative Genomics)是在基因组图谱和测序的基础上对已知的基因和基因组结构进行比较,来推测其他生物基因组的功能、表达机理和物种进化的学科。比较基因组学的基础是相关生物基因组的相似性。
基因组计划的进行推进了比较基因组学的发展,使得通过比对现有的EST、cDNA、mRNA 和基因序列数据来预测可变剪接成为可能。通过比较两种或多种基因组中同源外显子序列的相似性、可变剪接类型和相对表达量等信息,可以发现潜在的可变剪接变体[2]、判断可变剪接调控元件[3]或研究可变剪接外显子的进化保守性[4]。Ast 研究小组研究发现人类基因中保守的盒式外显子两侧的内含子序列在人和小鼠中一般也是保守的[5],他们提取了人类基因组中1996 个保守的外显子,通过分析在人和鼠中保守外显子序列的特征和人类基因中的外显子以及其相邻内含子在小鼠中同源序列的保守性,预测出人类基因中部分的盒式外显子[6-7]。Modrek 等人基于EST 比较了人与小鼠间可变剪接外显子的不同[4]。
比较基因组学的精确度取决于EST、cDNA、mRNA 数据源的可靠性,此方法对于具有物种特异性的可变剪接还是无法识别出来的。
1.2 基于高通量测序的可变剪接研究
高通量测序技术 (High-throughput sequencing)又称“下一代”测序技术,能一次并行对几十万到几百万条DNA 分子进行序列测定。自2005年454 Life Sciences 公司推出了454 FLX 焦磷酸测序平台之后,罗氏公司(Roche)、美国Illumina 公司和ABI 公司分别推出了454 测序仪(Roch GS FLX sequencer)、Solexa 基因组分析平台(Genome Analyzer platform)和SOLiD 测序仪 (ABI SOLiD sequencer),这三个测序平台即为目前高通量测序平台的代表,其测序技术比较见表1。
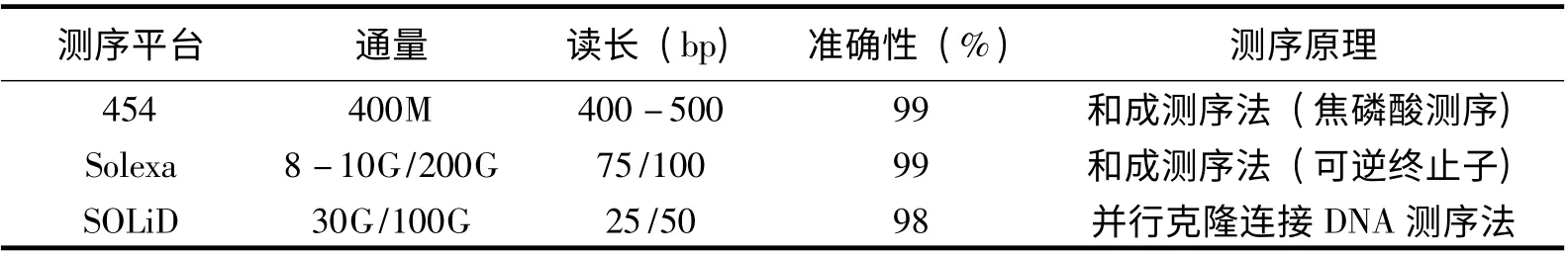
表1 各测序技术的比较
高通量测序在癌症中主要的关注对象是外显子组。Bradford 等人通过对SOLiD 和外显子芯片的结果进行分析比对发现两种方法在外显子表达量的测定上具有相同的效果[8]。Pan 等人用Solexa 测序方法对六种不同的正常的人组织进行测序,结果显示在约20%的多外显子基因中有新的可变剪接位点;通过对不同组织的结果进行比较发现大脑剪接水平的复杂度要高于其他的组织。[9]
采用微阵列等高通量技术必须先设计好探针,但是设计好的探针不能包括全部可能的剪接信息,所以高通量的测序方法不能完全满足实验研究的需求。
1.3 基于基因组序列特征的可变剪接研究
目前人们获得的基因组序列蕴含着大量的生物学信息,但是如何从这些信息中提取我们想要的有意义的信息是目前研究者们面临的难题。基于基因组序列特征研究使用模式识别算法、统计学方法和数学模型等方法对基因序列进行特征提取,使得实验数据能够从遗传物质本身出发提高了结果的说服力。
常见的识别方法有支持向量机(SVM),隐马氏模型(HMM),权重矩阵(weight matrix),神经网络模型(NN)等。Gideon Dror 等人利用盒式外显子和组成外显子的7 个特征构建了228 维的向量,用支持向量机对外显子进行分类识别。[10]Frey等人提取了1014 个基因组特征对盒式外显子进行分类预测[1]。邢永强等人分析老鼠盒式外显子和内含子保留型剪接位点附近的序列保守性特征,并用支持向量机和二次判别法对这两种剪接模式的供体端和受体端可变剪接位点进行了预测,取得较好的效果。[11]
可变剪接在真核生物机理中的重要性绝非一笔带过,研究其在生物疾病方面的调控对人类更全面的了解疾病产生的根源并且有效的控制疾病产生尤为重要,这里我们以可变剪接中盒式外显子为例做进一步的探讨。
2 盒式外显子的研究
盒式外显子(又称外显子跳跃)是指在某种剪接形式中一个外显子连同它两端的内含子一起被剪掉,从而使得该外显子不被表达。本文通过两种典型的疾病来说明盒式外显子的影响。
2.1 杜氏/贝氏肌营养不良
杜氏营养不良症(Duchenne Muscular Dystrophy)又称杜氏肌肉萎缩症,是一种X 染色体性联隐性遗传病,发病率约为活产男婴的1/3500,[12]为症状最严重的肌肉萎缩症。由于基因突变导致肌肉细胞不能正常产生一种称为Dystrophin 的蛋白质导致患者全身肌肉无力,其中大约65%的病例是由性染色体隐性遗传而来,35%则由基因突变引起。贝氏营养不良症(BMD)是一种病情较轻的肌肉萎缩症,是DMD 的亚型。由于目前医学上对DMD/BMD 尚无有效的治疗方法,所以从基因层面进行该疾病的筛选和预防不失为一个有效的方法。
在杜氏肌肉营养不良疾病中外显子缺失是很常见的,有文献指出外显子复制在基因突变中占有很大的比重(超过10%)[13],可以通过病人致病基因中外显子的多重跳跃来预防[12]。Caroline 等人[14]以患有肌肉萎缩症的金毛寻回犬为研究对象进行跟踪观察,成功地把以rAAV-U7 为中介的外显子跳跃方法运用到患有杜氏营养不良症的人的临床治疗中,并且达到了预期的效果。
2.2 脊髓性肌萎缩
脊髓性肌肉萎缩症(Spinal Muscular Atrophy)是常染色体隐性遗传病,是一种具有进行性、对称性、以近端为主的弛缓性瘫痪和肌肉萎缩为特征的遗传性下运动神经元疾病,该病的致病基因定位于Xq11-12[15]。人群发病 率为1/6000-1/10000,SMA 患儿存在SMN 端粒侧外显子7 的纯合性缺失。根据临床表现和发病时间,欧洲神经肌肉疾病中心将SMA 分为四种类型:SMAⅠ型(严重婴儿型,在出生后6 个月内发病)、AMAⅡ(迟发婴儿型,在出生后6-18 个月内发病)、SMAⅢ型(少年型,在出生18 个月后发病)和SMAⅣ型(成年型,一般30 岁后发病)。SMA 尚无特效治疗措施,进行产前基因诊断可以避免携带致病基因的胎儿出生。
SMN1 基因的缺失是SMA 疾病的发病机制,大约95%的SMA 患者存在SMN1 基因第7 号外显子的纯合性缺失[16]。Hui 等人[17]用计算方法模拟SMN1/2 上外显子7 的功能,通过对SMN1/2 上C6T,G-44A,A100G 和A215G4 个核苷酸的检测发现外显子7 的缺失主要是由C6T 引起的。孟英韬等人旨在探索将测序技术应用于缺失型脊髓性肌肉萎缩症基因诊断的可行性,其研究结果显示患者缺失SMN1 基因,缺失范围包括内含子6 至外显子8,结果与经典PCR-RFLP 方法的结果一致。
3 总结
可变剪接大量存在于人类基因组中,是蛋白质功能多样性的主要来源,也是杜氏营养不良、脊髓性肌肉萎缩症和癌症等疾病的发病机制之一。对可变剪接的研究方法在不断的提高,可变剪接数据库也在不断的更新丰富中。如何利用可变剪接的知识来诊断和预防疾病将会是研究者们重点研究的内容,主要的研究方向是:(1)融合基因组特征提高预测可变剪接事件的精度;(2)利用可变剪接与疾病产生之间的联系,构建针对特定疾病的可变剪接模型,进而对特定疾病起到更好的预测和诊断。
[1]Wang ZF,Burge CB. Splicing regulation:From a parts list of regulatory elements to an integrated splicing code[J].RNA,2008,14(1):802-813.
[2]Ohler U,Shomron N,Burge CB. Recognition of unknown conserved alternatively spliced exons[J].pLoS ComputBiol,2005,l(2):113-122.
[3]Fairbrother WG,Yeh RF,Sharp PA,et al. Predictive identification of exonic splicing enhancers in human genes[J].Science,2002,297(5583):1007-1013.
[4] Modrek B,Lee CJ. Alternative splicing in the human,mouse and rat genomes is associated with an increased rate of exon creation/loss[J].Nature,2003,34(2):177-180.
[5]Sorek R,Ast G. Intronic sequences flanking alternatively spliced exons are conserved between human and mouse[J].Genome Res,2003,13(7):1631 –1637.
[6]Sorek R,Shemesh R,Cohen Y,et al. A non-EST-based method for exon-skipping prediction[J].Genome Res,2004,14(8):1617-1623.
[7]Dror G,Sorek R,Shamir R. Accurate identification of alternatively spliced exons using support vector machine[J].Bioinformatics,2005,21(7):897-901.
[8]Bradford JR,Hey Y,Yates T Li Y,et al. A comparison of massively parallel nucleotide sequencing with oligonucleotide microarrays for global transcription profiling[J].BMC Genomics,2010,(11):641-648.
[9]Pan Q,Shai O,Lee LJ,et al. Deep surveying of alternative Splicing complexity in the human transcriptome by high-throughput sequencing[J].Nat Genet,2008,40(12):1413-1415.
[10]Gideon Dror,Rotem Sorek,Ron Shamir. Accurate identification of alternative spliced exons using support vector machine[J].Bioinformatics,2004,21(7):897-901.
[11]邢永强,张利绒. 老鼠基因组盒式外显子和内含子保留型可变剪接位点预测[J]. 内蒙古大学学报,2009,40(5):576-582.
[12]Echigoya Y,Yokota T. Skipping multiple exons of dystrophin transcripts using cocktail antisense oligonucleotides[J].Nucleic Acid Ther,2014,24(1):57-68.
[13]Kane L Greer,et al. Targeted Exon Skipping to Correct Exon Duplications in the Dystrophin Gen[J].Nucleic Acids,2014,3(155):1038-1044.
[14]CaroCaroline Le Guiner,et al. Forelimb Treatment in a Large Cohort of Dystrophic Dogs Supports Delivery of a Recombinant AAV for Exon Skipping in Duchenne Patients[J].Molecular Therapy,2014,11(22):1923-1935.
[15]Finsterer J. Perspectives of Kennedy’s disease[J].J Neurol Sci,2010,298(1-2):1-10.
[16]Lefebvre S,Burglen L,Reoullet S,et al. Identification and characterization of a spinal muscular atrophy-determining gene[J].Cell,1995,80(1):155-165.
[17]Hui Y,Xiong,et al. The human splicing code reveals new insights into the genetic determinants of disease[J].Science,2015,9(347):1-8.
猜你喜欢
国际太空(2023年1期)2023-02-27 09:03:42
内蒙古师范大学学报(自然科学汉文版)(2021年3期)2021-06-01 08:10:30
透析与人工器官(2020年1期)2020-11-16 01:42:34
航空学报(2020年4期)2020-06-08 01:37:34
铁道通信信号(2019年8期)2019-10-10 05:06:00
生物工程学报(2019年6期)2019-07-10 08:38:38
生物学通报(2019年1期)2019-02-15 16:33:43
生物学通报(2018年12期)2018-10-10 06:52:36
小演奏家(2018年5期)2018-09-04 09:52:48
中国发展观察(2017年8期)2017-04-26 03:51:50
