
Application of RP–HPLC method in dissolution testing and statistical evaluation by NASSAM for simultaneous estimation of tertiary combined dosages forms☆
2015-01-12 08:02YogeshUpadhyayNitinSharmaSarmaRavindraRawal
Yogesh Upadhyay,Nitin Sharma,G.S.Sarma,Ravindra K.Rawal
Department of Pharmaceutical Analysis,Indo-Soviet Friendship College of Pharmacy,Moga,Punjab 142001,India
Application of RP–HPLC method in dissolution testing and statistical evaluation by NASSAM for simultaneous estimation of tertiary combined dosages forms☆
Yogesh Upadhyay,Nitin Sharma,G.S.Sarma,Ravindra K.Rawal*
Department of Pharmaceutical Analysis,Indo-Soviet Friendship College of Pharmacy,Moga,Punjab 142001,India
A R T I C L E I N F O
Article history:
Received 5 May 2014
Received in revised form
2 November 2014
Accepted 18 November 2014
Available online 26 November 2014
Dissolution
RP–HPLC
Net analyte signal standard addition method
Two-way ANOVA
A dissolution method with robust high performance liquid chromatographic(HPLC)analysis for immediate release tablet formulation was developed and validated to meet the requirement as per International Conference on Harmonization(ICH)and United States Food and Drug Administration(USFDA) guidelines.The method involved the use of Agilent ZORBAX Eclipse XDB C18column,and temperature was maintained at 30°C.After optimization,the mobile phase was selected as phosphate buffer(KH2PO4, 30 mM)∶ACN(60∶40,v/v)with pH 3.0,and retention timeRtwas found as 3.24,4.16,and 2.55 min for paracetamol(PCM),chlorpheniramine maleate(CPM)and phenylephrine hydrochloride(PH)respectively at 265 nm and at a fow rate of 1 mL/min.The relative standard deviation(%RSD)for 6 replicate measurements was found to be less than 2%.Furthermore net analyte signal standard addition method (NASSAM)with spectrophotometer was performed for standard and liquid oral suspension.On the basis of selectivity,sensitivity and accuracy analysis,it was confrmed that this novel method could be useful for simultaneous estimation of the given drug combinations.Two-way analysis of variance(ANOVA)was applied for evaluating the statistical difference between the assay results obtained via both NASSAM and RP–HPLC methods and ultimately no signifcant difference was found between both the methods.All the methods and results were acceptable and confrmed that the method was suitable for intended use.
©2014 Xi'an Jiaotong University.Production and hosting by Elsevier B.V.All rights reserved.This is an open access article under the CC BY-NC-ND license(http∶//creativecommons.org/licenses/by-nc-nd/3.0/).
1.Introduction
High performance liquid chromatography(HPLC)method plays an important role in dissolution testing(DT)procedures.It provides a wide dynamic linear range,selectivity via separation and superior sensitivity.These features have been used to solve a variety of analytical problems encountered during DT of complex drug delivery systems.The linear range for an HPLC method occurs typically up to many orders of magnitude.The wide dynamic range often allows us to conduct the DT of formulation doses ranging from 0.1 to 200 mg with a single HPLC method.HPLC method also affords superior sensitivity over direct spectrophotometric method and is often used for DT of drug products with very low potencies[1].
The method has been validated to ensure that they are suitable for their intended use and give accurate and precise data.DT plays an important role in acquiring product sameness under scale up and post approval changes(SUPAC)related change.For solid dosage form,the characteristics of dissolution under physiological condition infuence in vitro dissolution.Solubility,permeability of drug products and release products(immediate/extended)are the major factors which affect the dissolution of development and quality control(QC)of synthetic as well as herbal drugs.The value of DT enhances signifcantly when performance of drug substance is evaluated as a function of time.DT is useful in QC and production batch to ensure similarities,so the DT remains similar and is crucial for clinical trial batches;further dissolution profling is used to support bioavailability and bioequivalence of a new pharmaceutical product[2].
This manuscript described the development and subsequent validation of methods via reverse phase high performance liquid chromatography(RP–HPLC)and net analyte signal standard addition method(NASSAM)and further applicability of the developed RP–HPLC method in DT of tablet formulation containing PCM,CPM and PH as active ingredients in combination.Method robustness is an essential parameter that should be studied and evaluated carefully[3].
In NASSAM,the part of the overlapping spectrum that is orthogonal to the space of other compounds(interferants)is known as NAS.It can be directly correlated to the analyte concentration in standard addition method.Therefore,the analyte concentrations can be determined simultaneously from a unique standard addition plot.NASSAM,as a new analysis method,is simple for estimating a drug with high precision and accuracy.It also requires no additional sample preparation.Hence,it can be a powerful and substituted method in comparison with HPLC for analysis of multiple components in simple steps[4].
Nowadays,NASSAM procedure has been widely applied alone or in conjugation with various sophisticated analytical methods, including DT of cocrystal forms[5].Other recent examples include simultaneous determination of sulphadiazine and trimethoprim in bovine milk and veterinary medicines[6],determination of sulfamethoxazole and trimethoprim in pharmaceutical formulations and biological f uids[7]and antazoline and naphazoline determination with NASSAM and spectrophotometric methods[8]. Simultaneous estimation is the analysis of standards present in multiple combination dosage form at the same time period.The advantages of simultaneous estimation lie in that it can avoid time consuming extraction and separation,and minimize the use of expensive reagents.And the method is further accurate and precise.
PCM,N-(4-hydroxyphenyl)acetamide[9](Fig.1),acts by inhibiting cyclo-oxygenase(COX-3,a linked variant of COX-1).It is an analgesic and antipyretic and used along with various cold preparations[10].
CPM,(3RS)-3-(4-Chlorophenyl)-N,N-dimethyl-3-(pyridin-2-yl) propan-1-amine hydrogen(Z)-butenedioate[9](Fig.1),is an antihistamine which has lower sedative effects than other antihistamines[10].
PH,(1R)-1-(3-hydroxyphenyl)-2-(methylamino)ethanol hydrochloride[9](Fig.1),is a selective α1-adrenergic receptor agonist used primarily in nasal decongestion,as an agent to dilate the pupil and to increase blood pressure[10].
All three drugs are of f cial in Indian pharmacopoeia(IP)[11]and British Pharmacopoeia(BP)[9].The PCM,CPM and PH alone or in combination with other drugs are reported to be estimated by the spectrophotometric method[12–15],derivative spectrophotometric method[16],chemometric-assisted spectrophotometry[17],HPLC [18,19],thin layer chromatography(TLC)[20],liquid chromatography–mass spectrometry(LC–MS)[21],Fourier transform infrared spectroscopy(FT–IR)[22],amperometric determination[23],f uorimetry[24], micellar electrokinetic chromatographic method[25],electrophoresis [26],liquid chromatography with two ultraviolet(UV)absorbance detectors[27],and chemometric determination[28].
Literature survey revealed that HPLC method was reported for this combination[29]but DT was not reported for tablets containing PCM,CPM and PH by robust RP–HPLC method.Furthermore,NASSAM was not available for this combination,which is a new,sensitive,economical and reliable analytical technique for simultaneous estimation of multicomponent mixtures.The present study mainly aimed at developing a DT procedure by developed RP–HPLC method for determination of PCM,CPM and PH in tablet dosage form,and further included analysis of signi f cant difference between the HPLC and NASSAM via two-way ANOVA.
2.Materials and methods
2.1.Materials
HPLC grade acetonitrile(ACN),potassium di-hydrogen phosphate buffer(KH2PO4),orthophosphoric acid(OPA)and hydrochloric acid(HCl)were purchased from Rankem(New Delhi,India).HPLC grade water for chromatography and dissolution[obtained from water puri f cation systems Milli-Q,ELIX 03(MILLIPORE,Milford,MA,USA)]was used.Solutions were f ltered through a qualisil nylon syringe f lter(25 mm×0.45 μm)Ultipor®N66®and membrane f lter(47 mm×0.45 μm)(Pall Pvt.Ltd.,India) prior to use.Standards of PCM,CPM and PH were procured from Syncom Health Care(Dehradun,India).Marketed formulation named as SNEEZY tablets was labeled as each uncoated tablet contained 500 mg of PCM,5 mg of PH and 2 mg of CPM(quinoline yellow)batch no.SC12030,manufactured by Cadila Pharmaceuticals,and COLD-GO which is an oral suspension was labeled as each 5 mL of oral suspension contained 125 mg of PCM,2 mg of CPM and 25 mg of PH(having coloring agent ponceau4R)batch no. HG-278,manufactured by Torque Pharmaceutical Limited,which was procured from local market(Moga,Punjab,India).
2.2.Instruments
HPLC system from WATERS(Milford,USA)is equipped with 515 HPLC pump as a solvent delivery system,rheodyne injection valve with a 20 μL loop and WATERS 2998 photodiode array(PDA)detector set at a wavelength range of 190–400 nm.Separation was performed on an Agilent ZORBAX Eclipse XDB“C18”column (4.6 mm×150 mm,5 μm).Chromatographic data were recorded and processed using EMPOWER-2 software.
Dissolution system from LABINDIA Disso 2000 is equipped with high precision multichannel pump and sample collector.For weighing analytical balance(Mettler Toledo and Sartorius)and pH measurement,pH meter(Mettler Toledo)was used.HPLC grade water was obtained from water puri f cation systems,Milli-Q,ELIX 03(MILLIPORE,Milford,MA,USA).
UV-vis double beam spectrophotometer Perkin-Elmer Lambda-35 was used for all spectrophotometric measurements(i.e.,for NASSAM),having a slit width of 1 nm,installed with UV-Winlab and UV-Winlab data processor and viewer software.All spectra were saved in comma separated f le(CSV)format and then data were statistically analyzed using unscramble 10.2.
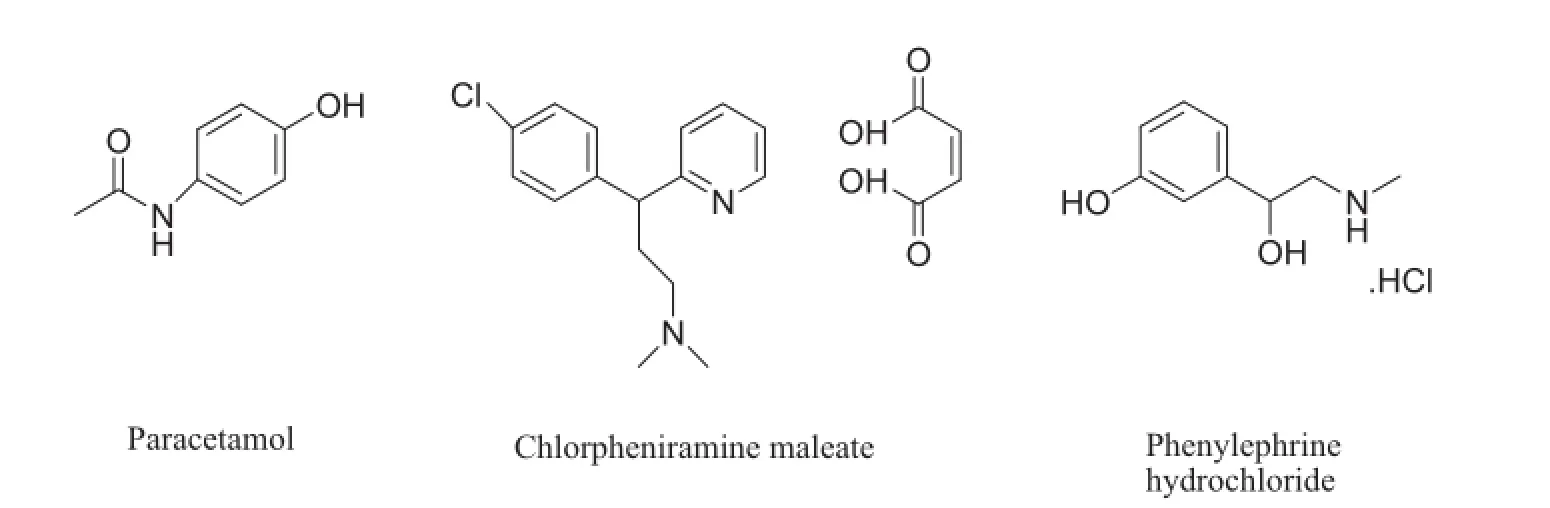
Fig.1.Chemical structures of paracetamol,chlorpheniramine maleate and phenylephrine hydrochloride.
2.3.Preparation of solutions
Pure samples stock solutions(1 mg/mL)of PCM,CPM and PH were freshly prepared in 0.1 M HCl,and further dilutions were made using mobile phase which was selected as 30 mM phosphate buffer(KH2PO4)∶ACN(60∶40,v/v),with pH adjusted to 3.0 with OPA.For solid dosage form analysis,20 tablets were weighed and triturated to obtain f ne powder.Stock solution was prepared in 0.1M HCl and further dilution was made with mobile phase. Standard addition method was performed in order to increase the concentration of CPM and PH in marketed formulation.The standard solutions of CPM and PH(10 μg/mL)were prepared by diluting the suitable aliquots of stock solution with 0.1 M HCl and further with mobile phase,and aliquots of spiking solution were spiked to marketed dilutions.For preparation of test samples(for oral suspension),solution was f rst extracted in 0.1 M HCl using sonication process for 1 h at room temperature.After this process, the resultant solution was f ltered through syringe f lter.Then further dilutions were made in mobile phase and processed for HPLC method.
For preparation of samples for NASSAM,stock solution was prepared in methanol,and further dilutions were made in diluting solvent,i.e.,methanol∶0.1 M HCl(1∶9,v/v).For preparation of samples for interference matrix,15 aliquots were prepared for PCM,CPM and PH(based on the linearity range for PCM,CPM and PH).Further the norms[i.e.,determination of sum of square of obtained data(X)and then square root of X component]were calculated by exporting the scanned spectra into CSV format.Similarly,15 aliquots of standard mixture were prepared as shown in Table 1.For calculating the interference matrices for PCM,mixture of CPM and PH was prepared within their linearity range,and the same procedures were applied for CPM and PH.For standard addition mixtures,tertiary mixture dilutions were prepared,keeping two drugs'concentrations constant on linearity basis.The same procedures were applied for CPM and PH.Finally we got 30 aliquots(15 for interference matrices and 15 for standard addition method matrices).
2.4.Methods
2.4.1.Optimization of chromatographic conditions
In order to achieve the best chromatographic separation,we changed different experimental variables.Finally,the appropriate conditions for method validation were selected.On the basis of system suitability parameters,i.e.,resolution factor(Rs),peak tailing factor(Tf),symmetry,retention time(Rt),capacity factor(k′) and height equivalent theoretical plates(HETP),the optimized chromatograms were selected for PCM,CPM and PH.The optimization parameters were signi f cantly affected by the mobile phase composition(type and composition of organic modi f ers/aqueous phase pH of solution,f ow rate,column temperature and wavelength).Further,these parameters were changed to achieve the best system suitability parameters.Various trials have been done in the above optimized parameters individually or in combination.To achieve the proper separation,various conditions were applied,which include mobile phase composition,i.e., phosphate buffer(KH2PO4)(20,25,30 and 35 mM)∶ACN in different ratios(55∶45,40∶60,60∶40,65∶35,45∶55 and 70∶30,v/v)at different pH(2,3 and 4),column temperatures(25,30 and 35°C), f ow rates(0.8,1 and 1.2 mL/min)and wavelengths(262,265 and 268 nm).
2.4.2.Effect of change in mobile phase composition
Different ratios of mobile phases(buffer KH2PO4and ACN) (55∶45,40∶60,60∶40,65∶35,45∶55 and 70∶30,v/v)and different molarities of aqueous phase(20,27,30 and 33 mM)were used. The optimized peak was resolved at(30 mM)phosphate buffer∶ACN(60∶40)with satisfactoryRs,Tf,HETP and symmetry.In another composition we found variable deviations from standard value for all SST parameters as shown in Fig.2(C),(F),and(I).
2.4.3.Effect of change in pH
Optimizations were performed by varying the pH(2.7,3 and 3.3)of aqueous phase(KH2PO4),while the other factors were kept constant.Number of theoretical plates(n),Rs,k′,Tfand HETP were found optimum for method development at 3 pH.TheRswas decreased for both PCM and CPM with increase in HETP at pH 2.7 and 3.3 as shown in Fig.2(B),(E),and(H).
2.4.4.Effect of change infow rates
Various f ow rates were tried(0.8,1 and 1.2 mL/min)that affected theRtof drugs.By increasing the f ow rate,theRtwas decreased and vice versa.HETP was found highest at 1.2 and 0.8 mL/ min.At 0.8 mL/min,Tfwas found to be>2 for PCM.Finally the f ow rate 1 mL/min was selected,while other parameters were kept constant.Effects of f ow rates for PCM,CPM and PH are shown in Fig.2(A),(D),and(G).
2.4.5.Effect of change in column oven temperatures
Various column oven temperatures(27,30 and 33°C)were employed.At 27°C,merging of PCM and CPM occurred withRs<2,and at 33°C,peak broadening and increase in HETP value occurred for CPM.Finally,30°C was selected as optimized column oven temperature for study,at which all SST parameters were found to be superlative.
2.5.Finalized chromatographic conditions after optimization
After analyzing all robustness parameters and optimization conditions,the recommended condition was the mobile phase consisting of phosphate buffer(KH2PO4,30 mM)∶ACN(60∶40,v/v) with pH 3.The chromatograms after optimization showedsymmetric and sharp peak with theRtof 3.24,4.16 and 2.55 min for PCM,CPM and PH respectively as shown in Fig.3.The best resolution and sensitivity of the method were obtained at 265 nm at a f ow rate of 1 mL/min and column oven temperature of 30°C.
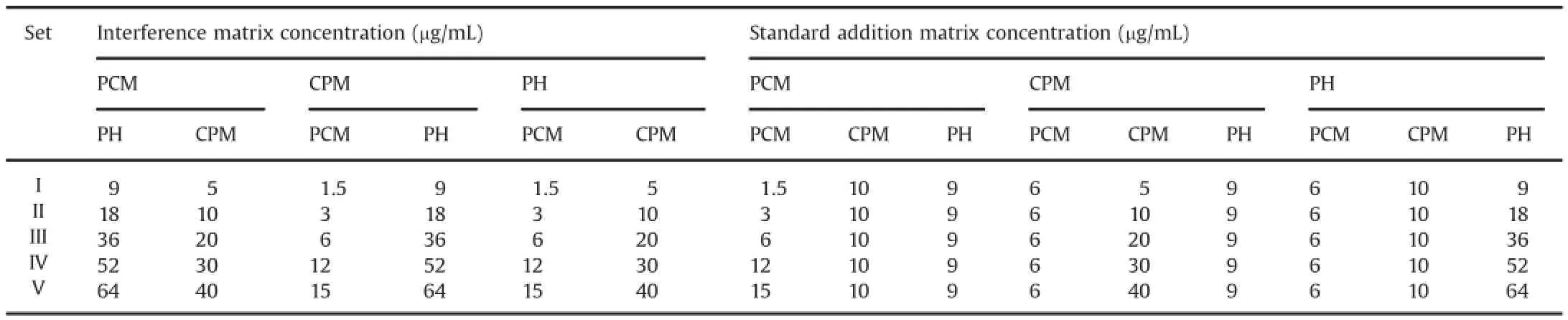
Table 1Aliquots concentrations of all three drugs used in interference matrix and standard addition matrix.
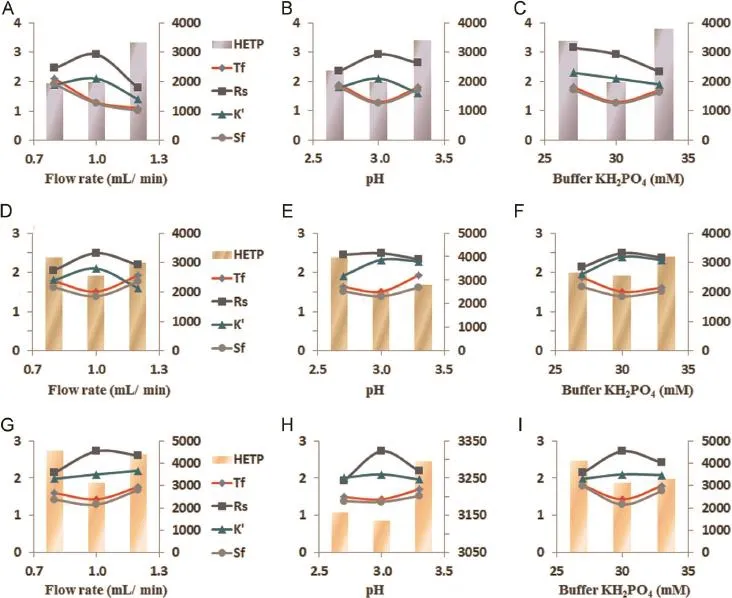
Fig.2.Optimization graphs for PCM(A,B and C),CPM(D,E and F)and PH(G,H and I)in whichxaxis indicates the f ow rate(mL/min),pH and buffer molarity(mM). Primaryyaxis indicates tailing factor(Tf),resolution(Rs),capacity factor(K′),symmetric factor(Sf),and secondaryyaxis indicates height equivalent theoretical plates(HETP).
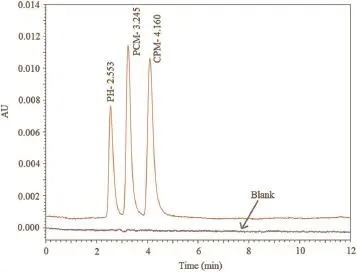
Fig.3.Chromatogram of PCM,CPM and PH after f nal optimization.
2.6.Dissolution test(DT)conditions
The DT was performed in compliance with United States Pharmacopoeia(USP)(711)using apparatus 2 with paddles.For optimization of medium and paddle,speeding of a dissolution medium/agitation screening was performed.After optimization, medium was selected as 0.1 M HCl(having a pH of 1.8 gastrointestinal tract).Because the tablets were uncoated and disintegrated immediately in gastrointestinal tract,0.1 M HCl was selected as a dissolution medium.The performances of dissolution apparatuses are highly dependent on hydrodynamics which includes coning and may affect the dissolution study.In order to minimize coning,paddle speed was optimized as 50 rpm.Media volume of 900 mL was f lled in six baskets and two baskets were used as blank for replenishing.The medium,before processing, was degassed via sonication process,and temperature was set at 37±0.5°C.At different time intervals(0–150 min),samples were drawn off(n=6,samples were drawn off at each time interval). Due to the immediate releasing property of these tablets,the earlier time intervals provided more distinguished ability.Autosamplings were performed and samples were f ltered through syringe f lters.
2.7.HPLC method
Method validation was performed after getting optimized peak. Isocratic elution was done using 1 mL/min f ow rate with phosphate buffer(KH2PO4,30 mM)∶ACN(60∶40,v/v)at pH 3(adjusted with OPA).Fresh mobile phases were prepared for each analysis. Before introducing into the system,the mobile phase was f ltered through 0.45 μm membrane f lters and degassed through sonicator.Pre-UV scanning(380–180)was done and f nal wavelength was selected as 265 nm for all estimations,because UV detection in this wavelength provided the optimal sensitivity needed for excellent quanti f cation of the low drug concentration of marketed formulation.To achieve the equilibrium,the column was saturated at least 30 min before the analysis.The substances were quanti f ed using peak area ratio.
2.8.NASSAM method
NAS technique is de f ned as the net analyte signal for an analyte as a part of its spectrum which is orthogonal to the space spanned by the spectra of all other analytes.It is given by the following equation[4,30]∶

whereYdenotes theI×Jmatrix having calibration response ofIsamples atJsensors,Ais the spectrum of given sample(spectrumCkof purekat unit concentration)and number of spectra used to build the model,Dis the J×J identity matrix,Ykis aJ×Lcolumn spaced spanned by the spectra of all other analytes except thatk[(Yk)+is the pseudo inverse ofYk,Lis the number of spectral factors used to build the model],andBNAS,kis theJ×JNAS space.
The standard addition method was used to eliminate the calibration and prediction steps of multivariate calibrations method and determination was carried out in a single step for each analyte.For simultaneous estimation by this technique,it required spectrum vector of mixtures.The standard concentrations of PCM, CPM and PH were simultaneously added to the sample solutions. The spectrum was recorded after each standard addition based on the following equations∶
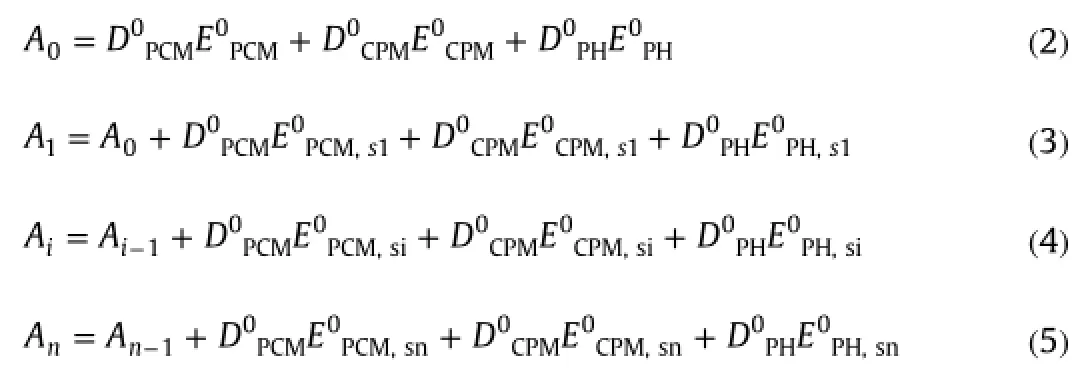
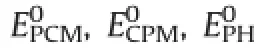
NAS vector(Fig.4)for PCM,CPM and PH compounds after each standard addition,NASPCM,NASCPM,and NASPHcan be found from the following equation,respectively∶

whereIdenotes identical matrix,R,SandTare the matrices of absorbances at different concentrations of both interferences according to Table 1.InR+,S+,andT+,the superscript(+)denotes the pseudoinverse ofR,S,andTmatrices respectively.The spectrum of mixtureAiis a combination of two independent parts NASPCM,orthogonal to space of interferences(CPM and PH)and (R+R),Aiis generated by linear combination of the spectra of interfering agents and it denotes standard addition matrix as shown in Table 1.
Fig.4.Vector space for analyte(PCM)and other analytes(CPM and PH)in two dimensions.
3.Results and discussion
3.1.Validation
Linearity,range,accuracy,robustness,limit of detection(LOD), limit of quanti f cation(LOQ),selectivity and sensitivity were useful validation parameters for comparison of methods as well as for determination of quality of given spectral analytical techniques[3].3.1.1.Calibration and linearity
HPLC standard mixtures were prepared for all three drugs in their linearity range,and calibration curve was plotted.Samples were injected in triplicate(validation parameters are shown in Table 2).The retention time of standards was 3.24 min for PCM, 4.16 min for CPM and 2.55 min for PH.A typical HPLC chromatogram of the standard mixtures is shown in Fig.3.Then peak area against the concentration of the drugs was plotted to obtain the calibration graphs.These calibration graphs were found to be linear in the stated range.For NASSAM the calibration graphs were obtained by calculating norms of interference matrix data sets.For NASSAM and RP–HPLC methods correlation coef f cients for PCM, CPM and PH were found to be 0.997<R2<1.Overlay chromatogram and calibration curve obtained via HPLC methods are shown in Fig.5.NAS overlay spectra for PCM,CPM and PH are shown in Figs.6A,7A,and 8A,respectively.
3.1.2.Precision
The inter-day and intra-day precision parameters were studied for all the drugs(n=3).The percent relative standard deviation was calculated,which was found to be less than 2%for all drugs, indicating that the method was reliable and reproducible.The precision data are shown in Table 3.
3.1.3.Assay and recovery studies
The recovery tests were performed by adding the known amount of each standard drug in suspension as well as tablets.Thesame procedure was applied for NASSAM for estimating suspension.Results having excellent recoveries were obtained.Assay of marketed formulation was done and is shown in Table 4.Recoverystudies for suspension and tablets are shown in Table 5 and Table 6,respectively,and the concentrations shown in Table 4 and Table 5wereobtainedaftersubtractingthestandardadded concentrations.
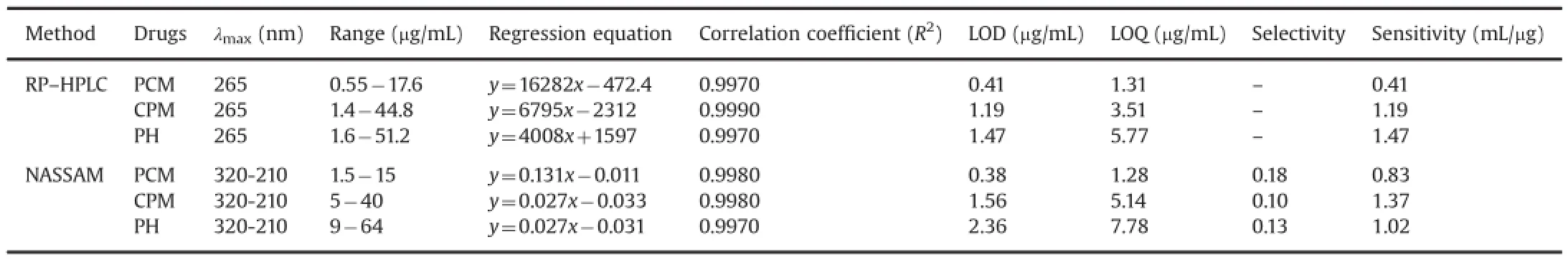
Table 2Validation parameters obtained by all methods.

Fig.5.Overlay chromatogram of PCM,CPM and PH.
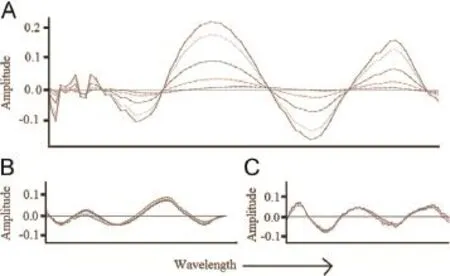
Fig.6.(A)NAS spectrum for PCM,(B)overlapped spectra for CPM and(C)overlapped spectra for PH.

Fig.7.(A)NAS spectrum for CPM,(B)overlapped spectra for PH,and(C)overlapped spectra for PCM.
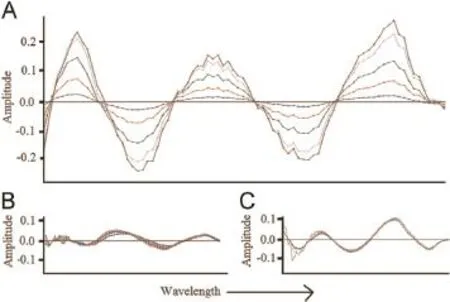
Fig.8.(A)NAS spectrum for PH,(B)overlapped spectra for PCM,and(C)overlapped spectra for CPM.

Table 3Precision data obtained via RP–HPLC method for all drugs.

Table 4Assay of marketed formulations.
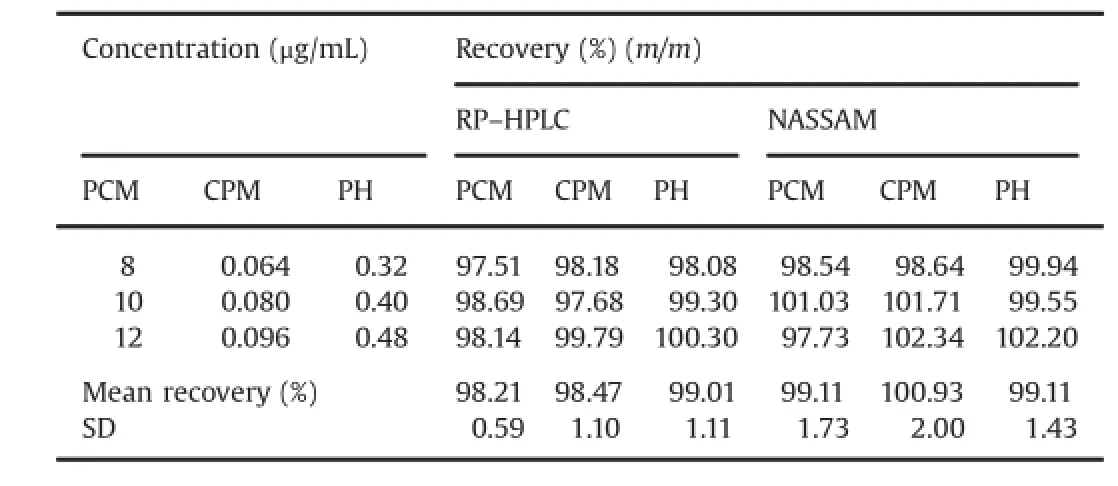
Table 5Recovery studies results for oral suspension dosage form by RP–HPLC and NASSAM.

Table 6Recovery studies results for tablet dosage form by RP–HPLC method.
3.1.4.LOD and LOQ
LOD was the concentration of analyte that produces analytical signal equal to thrice the deviation of background signals.The LOQ was the lowest amount of analyte which could be quantifed. These parameters were studied for both methods.
For NASSAM,LOD is estimated by the following equation∶

whereXdenotes the measure of measurement errors.This can be estimated by calculating the norms of several blank samples (n=10).NASTiis the net analyte signal of T component[4,30,31]. Data obtained via NASSAM and RP–HPLC methods are shown in Table 2.
3.1.5.Selectivity and sensitivity
In simple defnition selectivity refers to a method that produces a response of a single analyte.No interference was observed,resulting from the substance present in formulation.Sensitivity refers to as the response due to a particular analyte varies as a function of its concentration.In RP–HPLC method,one can directly select the sensitivity value equivalent to LOD value because as per defnition sensitivity is also known as minimum detectable concentration of analyte.But for NASSAM,we can easily calculate the sensitivity of the analyte using Eq.(10),and due to a large number of spectral data and human errors,sometimes,sensitivity values are deviated from LOD value as shown in Table 2.
Orthogonal spectra refers to spectra with no overlap had selectivity equal to 1,whereas identical spectra have selectivity equal to zero[4,30,31].Practically,in NASSAM,we have a large number of test samples,which lead to human errors as well,and for this reason,selectivity is not always equal to zero.As suggested by Garner[32],selectivity and sensitivity were calculated in two different ways,i.e.multivariate fgures of merit by using analytical blanks in which 10 different blanks of diluting solvent were scanned,and analytical signals obtained were used to generate the NAS norms;the second method includes use of NAS obtained from samples with low concentration,fxing near to the lower limit of the linear range and variable concentrations of the other ones in all the ranges shown in the calibration sets.
By NASSAM,these parameters are calculated as

Table 7System suitability parameters for all drugs.

where NASTiis the net analyte signal of T component and Ti denotes the total signal of T components[31].Data for these parameters are shown in Table 2.
3.1.6.System suitability parameters
The system suitability parameters were estimated for precise method development.These parameters were found within the specifed limits and are shown in Table 7.
3.1.7.Robustness and ruggedness
The factors which affect the peak symmetry are percentage organic/aqueous mobile phase,fow rate,temperature and wavelength.For this study,the symmetry was found to be consistently less than 1.64 across all the studies.So the suitability of method did not affect with respect to peak symmetry.Further,the 10% variations were done for all parameters as shown in Fig.2.The fow rates and%organic phases affected the peak effciency and retention time.The aim of robustness study was to fnd an actual value for organic phase composition,fow rate,pH and temperature for this method.
Ruggedness is essential to performing as per USFDA guidelines. It is defned as reproducibility of an analytical method obtained by the analysis of same sample under different variable conditions like different laboratories,analysts,instruments,and environmental conditions.The test results obtained in terms of%RSD were found to be less than 2%.Results obtained via robustness and ruggedness studies are shown in Table 8.
3.1.8.Percentage drug release
At different time intervals[0,5,10,15,30,60,90,120 and 150 min(n=6,samples were drawn off at each time interval)],the release rate of tablet dosage form having PCM,CPM and PH was noted.Retention time was found to be 3.24,4.16 and 2.55 min for PCM,CPM and PH,respectively.Overlay chromatogram obtained at various time intervals is shown in Fig.9.Data obtained with the approximately same retention time were found excellent percentage drug release rate and are shown in Table 9.
3.2.Signifcant difference between RP–HPLC and NASSAM via twoway ANOVA
Two-way ANOVA is an appropriate analysis method for a study with a quantitative outcome of two(or more)categorical explanatory variables.Here we have applied this statistical techniquein the assay data obtained via both methods(RP–HPLC and NASSAM)for combined oral suspension as shown in Table 10.Results were satisfactory and found within the limits.Calculated values were less than the theoretical value.At 0.05 level of signifcanceP-value was found to be less than 0.05 andF-value was found to be less thanF-critical value at the same signifcance level of analysis, which indicates that there is no signifcant difference between both the methods.Two-way ANOVA results obtained via comparing assay data of RP–HPLC and NASSAM for oral suspension are shown in Table 11.
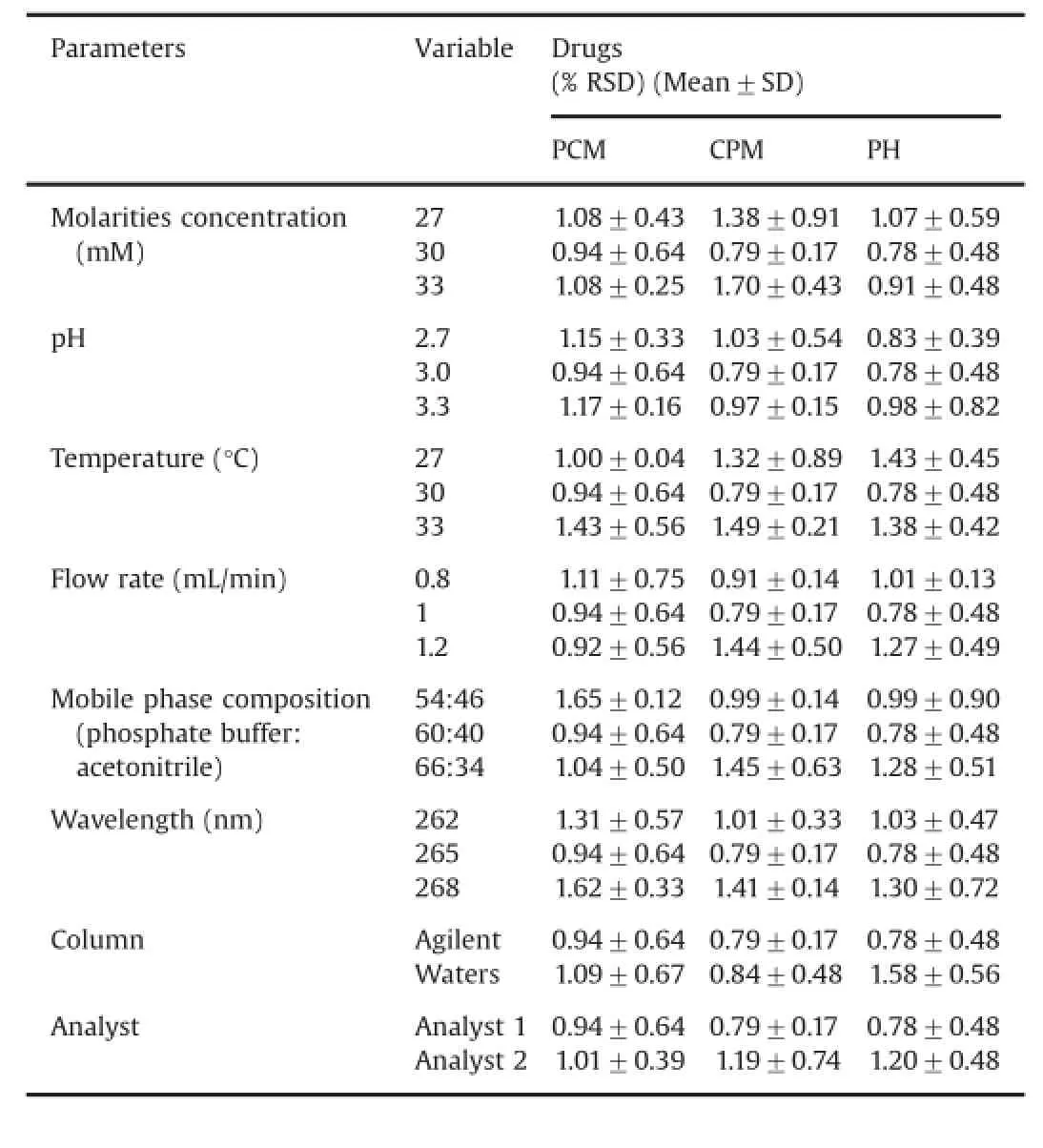
Table 8Robustness and ruggedness data obtained from RP–HPLC method for all drugs.
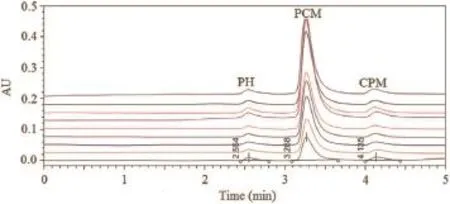
Fig.9.Overlay chromatogram obtained via dissolution method at various time intervals.

Table 9Percentage drug release at different time intervals for PCM,CPM and PH.
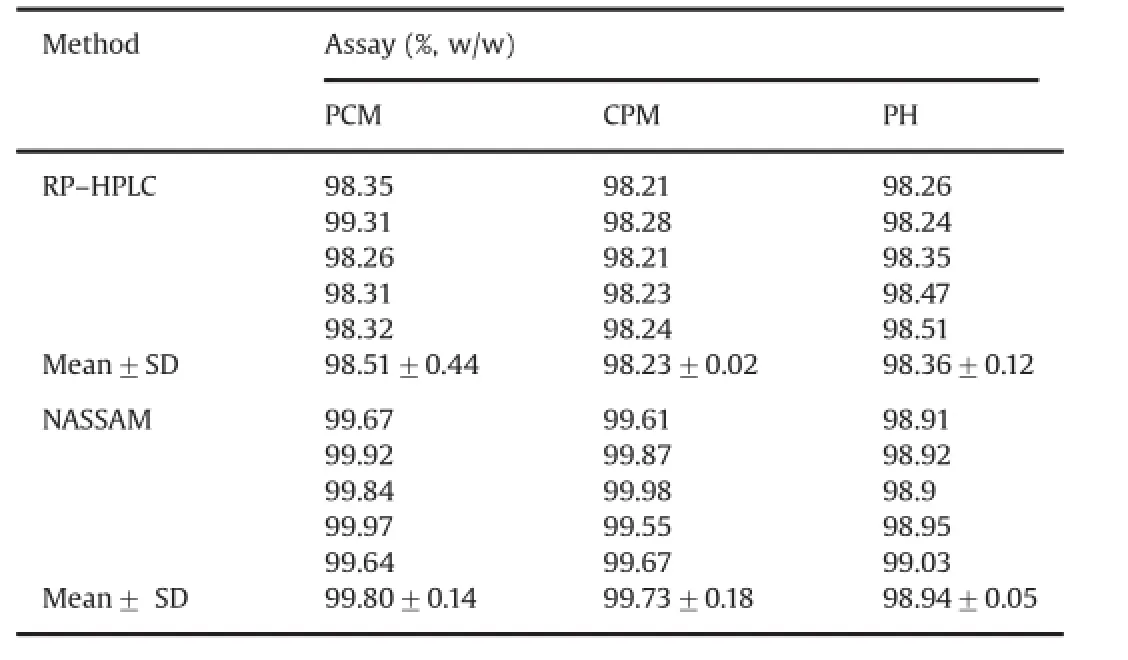
Table 10Assay data used in two-way ANOVA for statistical comparison between RP–HPLC and NASSAM for oral suspension.
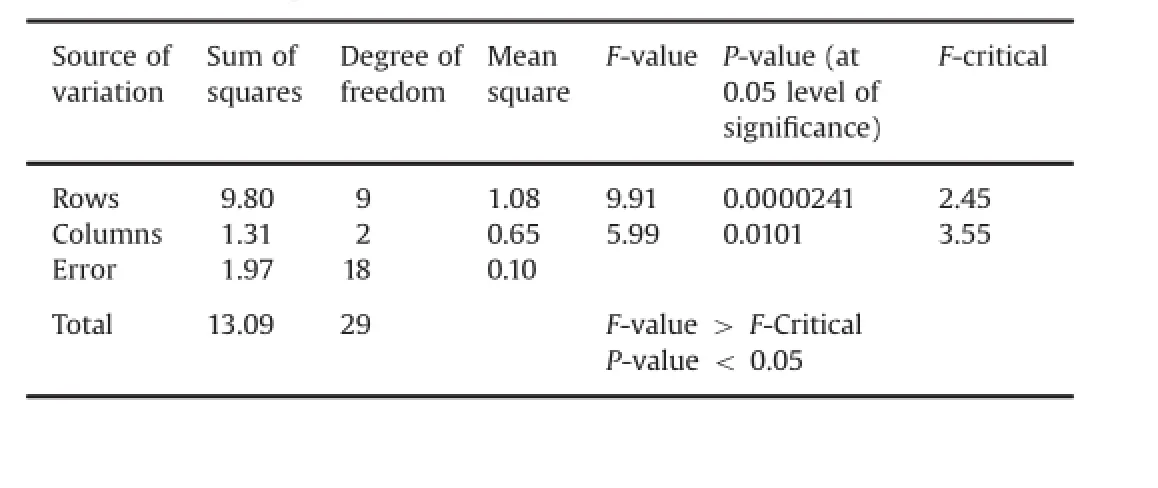
Table 11Two-way ANOVA results obtained via comparing assay data of RP–HPLC and NASSAM for oral suspension.
4.Conclusion
The optimization of RP–HPLC method showed that the mobile phase composition,pH and fow rate were more crucial parameters to be controlled for reproducible and quantitative estimation of PCM,CPM and PH.Simple and reproducible sample extraction for oral suspension provided higher sensitivity for determination of tertiary combination of drugs by RP–HPLC and NASSAM.The developed RP–HPLC method could further be applicable to bio-analytical method development,stability indicating method or in vitro–in vivo correlation studies for this particular combination.
The validated developed RP–HPLC and NASSAM were found simple,specifc,accurate,rapid,precise,economical and reliable for contemporary analysis of drugs in tablet form as well as oral suspension.Validation parameters for this particular situation were adequate for both methods.Dissolution procedure was performed to characterize the drug release rate for tablet dosage form and further analyzed by developed RP–HPLC method.Successfully two-way ANOVA was applied(in assay results)between RP–HPLC and NASSAM for analysis of oral suspension.Further,it was concluded that there was no signifcant difference between both the methods.The dissolution and simultaneous estimation procedure could be applied to routine quality control analysis of oral suspension as well as solid dosage form.
[1]Q.Wang,D.Ma,J.P.Higgins,Analytical method selection for drug product dissolution testing,Dissolution Technol.24(2006)6–13.
[2]J.S.Space,A.M.Opio,B.Nickerson,et al.,Validation of a dissolution method with HPLC analysis for lasofoxifene tartrate low dose tablets,J.Pharm.Biomed. Anal.44(2007)1064–1071.
[3]R.A.Nash,A.H.Wachter,Pharmaceutical Process Validation,Marcel Dekkar, Inc.,New York,2003,pp.507–519.
[4]R.Hajian,A.Karamian,The use of net analyte signal aspect in univariate calibration for simultaneous determination of guaiphenesin,pseudoephedrine and chlorpheniramine in cough syrup formulations,Curr.Pharm.Anal.8 (2012)93–100.
[5]A.Shayanfar,K.Asadpour-Zeynali,A.Jouyban,Solubility and dissolution rate of a carbamazepine–cinnamic acid cocrystal,J.Mol.Liq.187(2013)171–176.
[6]R.Hajian,E.Mousavi,N.Shams,Net analyte signal standard addition method for simultaneous determination of sulphadiazine and trimethoprim in bovine milk and veterinary medicines,Food Chem.138(2013)745–749.
[7]M.H.Givianrad,M.Mohagheghian,Net analyte signal standard additions method for simultaneous determination of sulfamethoxazole and trimethoprim in pharmaceutical formulations and biological f uids,E-J.Chem.9(2012) 680–692.
[8]K.Asadpour-Zeynali,R.Ghavami,R.Esfandiari,et al.,Simultaneous determination of antazoline and naphazoline by the net analyte signal standard addition method and spectrophotometric technique,J.AOAC Int.93(2010) 1995–2001.
[9]British Pharmacopoeia,The Department of Health∶British Pharmacopoeia Commission,The Stationery Of f ce,London,2009,p.147,483,503.
[10]H.P.Rang,M.M.Dale,J.M.Ritter,et al.,Rang and Dale's Pharmacology, Churchill Livingstone Elsevier,New York,2007,p.186,236,364.
[11]Indian Pharmacopoeia,Government of India∶Ministry of Health and Family Welfare,Indian Pharmacopoeia Commission,Ghaziabad,2007,p.301,900, 936.
[12]E.Dinç,C.Yücesoy,F.Onur,Simultaneous spectrophotometric determination of mefenamic acid and paracetamol in a pharmaceutical preparation using ratio spectra derivative spectrophotometry and chemometric methods,J. Pharm.Biomed.Anal.28(2002)1091–1095.
[13]I.Savić,G.Nikolić,V.Bankovic,Development and validation of spectrophotometric method for phenylephrine hydrochloride estimation in nasal drops formulations,Maced.J.Chem.Chem.Eng.27(2008)149–156.
[14]M.Nogowska,I.Muszalska,M.Zajac,Simultaneous spectrophotometric determination of acetylsalicylic acid,paracetamol and caffeine in pharmaceutical preparations,Chem.Anal.(Warsaw)44(1999)1041–1048.
[15]S.S.Chitlange,R.Soni,S.B.Wankhede,et al.,Spectrophotometric methods for simultaneous estimation of dexibuprofen and paracetamol,Asian J.Res.Chem. 2(2009)30–33.
[16]P.R.Kumar,M.M.Krishna,P.B.Prakash,et al.,Derivative spectrophotometric estimation of ondansetron and paracetamol,J.Chem.3(2006)134–141.
[17]N.H.Al-Shaalan,Determination of phenylephrine hydrochloride and chlorpheniramine maleate in binary mixture using chemometric-assisted spectrophotometric and high-performance liquid chromatographic-UV methods,J. Saudi Chem.Soc.14(2010)15–21.
[18]G.W.Schieffer,D.E.Hughes,Simultaneous stability indicating determination of phenylephrine hydrochloride,phenylpropanolamine hydrochloride and guaifenesin in dosage forms by reversed phase paired ion high performance liquid chromatography,J.Pharm.Sci.72(1983)55–59.
[19]R.Ragonese,M.Mulholland,J.Kalman,et al.,Full and fractionated experimental designs for robustness testing in the high performance liquid chromatographic analysis of codeine phosphate,pseudoephedrine hydrochloride and chlorpheniramine maleate in a pharmaceutical preparation,J.Chromatogr.A 870(2000)45–51.
[20]A.P.Argekar,J.G.Sawant,Simultaneous determination of paracetamol and mefenamic acid in tablets by HPTLC,J.Planar.Chromatogr.Mod.TLC 12(1999) 361–365.
[21]M.Godejohann,L.H.Tseng,U.Braumann,et al.,Characterization of a paracetamol metabolite using on-line LC–SPE–NMR–MS and a cryogenic NMR probe,J.Chromatogr.A 1058(2004)191–196.
[22]S.Ekgasit,N.Pattayakorn,D.Tongsakul,et al.,A novel ATR FT-IR microspectroscopy technique for surface contamination analysis without interference of the substrate,Anal.Sci.23(2007)863–868.
[23]P.S.J.Richard,N.S.Sriman,Amperometric determination of hydrazine using a surface modi f ed nickel hexacyanoferrate graphite electrode fabricated following a new approach,J.Electroanal.Chem.617(2008) 111–117.
[24]E.J.Llorent-Martinez,D.Šatínský,P.Solich,et al.,Fluorimetric SIA optosensing in pharmaceutical analysis∶determination of paracetamol,J.Pharm.Biomed. Anal.45(2007)318–321.
[25]D.Emre,N.Özaltın,Simultaneous determination of paracetamol,caffeine and propyphenazone in ternary mixtures by micellar electrokinetic capillary chromatography,J.Chromatogr.B 847(2007)126–132.
[26]M.R.Gomez,R.A.Olsina,L.D.Martinez,et al.,Simultaneous determination of dextromethorphan,diphenhydramine and phenylephrine in expectorant and decongestant syrups by capillary electrophoresis,J.Pharm.Biomed.Anal.30 (2002)791–797.
[27]U.R.Cieri,Determination of phenylephrine hydrochloride,chlorpheniramine maleate and methscopolamine nitrate in tablets or capsules by liquid chromatography with two UV absorbance detectors in series,J.AOAC Int.89 (2006)53–57.
[28]M.R.Khoshayand,H.Abdollahi,A.Ghaffari,et al.,Simultaneous spectrophotometric determination of paracetamol,phenylephrine and chlropheniramine in pharmaceuticals using chemometric approaches,J.Fac.Pharm.18 (2010)292–297.
[29]H.Senyuva,T.Ozden,Simultaneous high-performance liquid chromatographic determination of paracetamol,phenylephrine hydrochloride,and chlorpheniramine maleate in pharmaceutical dosage forms,J.Chromatogr.Sci.40 (2002)97–100.
[30]K.Asadpour-Zeynali,M.Bastami,Net analyte signal standard addition method (NASSAM)as a novel spectro f uorimetric and spectrophotometric technique for simultaneous determination,application to assay of melatonin and pyridoxine,Mol.Biomol.Spectrosc.75(2010)589–597.
[31]M.M.Galera,D.P.Zamora,J.L.M.Vidal,Determination of carbendazim,thiabendazole and fuberidazole using a net analyte signal-based method,Talanta 59(2003)1107–1116.
[32]F.C.Garner,G.L.Robertson,Evaluation of detection limit estimators,Chemometr.Intell.Lab.3(1988)53–59.
☆Peer review under responsibility of Xi'an Jiaotong University.
*Corresponding author.Tel.∶+91 16 3632 4200;fax∶+91 16 3623 9515.
E-mail addresses:ganti_ss@rediffmail.com(G.S.Sarma),
rawal.ravindra@gmail.com(R.K.Rawal).
http∶//dx.doi.org/10.1016/j.jpha.2014.11.001
2095-1779/©2014 Xi'an Jiaotong University.Production and hosting by Elsevier B.V.All rights reserved.This is an open access article under the CC BY-NC-ND license (http∶//creativecommons.org/licenses/by-nc-nd/3.0/).
 Journal of Pharmaceutical Analysis2015年5期
Journal of Pharmaceutical Analysis2015年5期
- Journal of Pharmaceutical Analysis的其它文章
- Species authentication and geographical origin discrimination of herbal medicines by near infrared spectroscopy∶A review☆
- Four new degradation products of doxorubicin∶An application of forced degradation study and hyphenated chromatographic techniques☆
- Multiple responses optimization in the development of a headspace gas chromatography method for the determination of residual solvents in pharmaceuticals☆
- Determination of atractylon in rat plasma by a GC–MS method and its application to a pharmacokinetic study☆
- Rapid screening and distribution of bioactive compounds in different parts ofBerberis petiolarisusing direct analysis in real time mass spectrometry☆
- High-sensitivity simultaneous liquid chromatography–tandem mass spectrometry assay of ethinyl estradiol and levonorgestrel in human plasma☆