
High-sensitivity simultaneous liquid chromatography–tandem mass spectrometry assay of ethinyl estradiol and levonorgestrel in human plasma☆
2015-01-12 08:02:42AbhishekGndhiSwtiGuttikrPritiTrivedi
Abhishek Gndhi,Swti Guttikr,Priti Trivedi
aBioanalytical Research,Veeda Clinical Research,Ahmedabad 380059,India
bDepartment of Pharmaceutical Chemistry,K.B.Institute of Pharmaceutical Education and Research,Kadi Sarva Vishawavidyalaya, Gandhinagar 382023,India
High-sensitivity simultaneous liquid chromatography–tandem mass spectrometry assay of ethinyl estradiol and levonorgestrel in human plasma☆
Abhishek Gandhia,b,Swati Guttikara,Priti Trivedib,*
aBioanalytical Research,Veeda Clinical Research,Ahmedabad 380059,India
bDepartment of Pharmaceutical Chemistry,K.B.Institute of Pharmaceutical Education and Research,Kadi Sarva Vishawavidyalaya, Gandhinagar 382023,India
A R T I C L E I N F O
Article history:
Received 4 September 2014
Received in revised form
15 January 2015
Accepted 12 February 2015
Available online 20 February 2015
Ethinyl estradiol
Levonorgestrel
LC–MS/MS
Human plasma
Derivatization
A sensitive and simultaneous liquid chromatography–tandem mass spectrometry method was developed and validated for quantifcation of ethinyl estradiol and levonorgestrel.The analytes were extracted with methyl-tert-butyl ether∶n-hexane(50∶50,v/v)solvent mixture,followed by dansyl derivatization.The chromatographic separation was performed on a Kinetex C18(50 mm×4.6 mm,2.6 μm)column with a mobile phase of 0.1%(v/v)formic acid in water and acetonitrile in gradient composition.The mass transitions were monitored in electrospray positive ionization mode.The assay exhibited a linear range of 0.100–20.0 ng/mL for levonorgestrel and 4.00–500 pg/mL for ethinyl estradiol in human plasma.A run time of 9.0 min for each sample made it possible to analyze a throughput of more than 100 samples per day.The validated method has been successfully used to analyze human plasma samples for application in pharmacokinetic and bioequivalence studies.
©2015 Xi'an Jiaotong University.Production and hosting by Elsevier B.V.All rights reserved.This is an open access article under the CC BY-NC-ND license(http∶//creativecommons.org/licenses/by-nc-nd/4.0/).
1.Introduction
Levonorgestrel/ethinyl estradiol is a progesterone and estrogen combination birth control pill.It works by preventing ovulation, thickening the mucus in the cervix and changing the lining of the uterus[1].Combination oral contraceptives act by suppression of gonadotropins[2].
Levonorgestrel is rapidly and completely absorbed after oral administration(bioavailability about 100%).It is not subjected to frstpass metabolism or enterohepatic circulation.Therefore,it does not undergo variations in absorption after oral administration.Ethinyl estradiol is rapidly and almost completely absorbed by the gastrointestinal tract,but due to frst-pass metabolism in gut mucosa and liver,the bioavailability of ethinyl estradiol is within 38%–48%[2].
The kinetics of total levonorgestrel is non-linear due to an increase in binding of levonorgestrel to sex hormone binding globulin(SHBG),which is attributed to increased SHBG level that is induced by the daily administration of ethinyl estradiol[2].Levonorgestrel in serum is primarily bound to SHBG.Ethinyl estradiol is about 97%bound to plasma albumin.Ethinyl estradiol does not bind to SHBG,but induces SHBG synthesis.
☆
The most important metabolic pathway of levonorgestrel occurs in the reduction of the D 4-3-oxo group and hydroxylation at positions 2a,1b,and 16b,followed by conjugation.Most of the metabolites that circulate in the blood are sulfates of 3a,5b-tetrahydro-levonorgestrel,while excretion occurs predominantly in the form of glucuronides.Some of the parent levonorgestrel also circulates as 17b-sulfate[2].
Cytochrome P450 enzymes(CYP3A4)in the liver are responsible for the 2-hydroxylation of ethinyl estradiol,the major oxidative reaction.The 2-hydroxy metabolite is further transformed by methylation and glucuronidation prior to urinary and fecal excretion.Levels of cytochrome P450(CYP3A)vary widely among individuals and can explain the variation in rates of ethinyl estradiol 2-hydroxylation.Ethinyl estradiol is excreted into the urine and feces as glucuronide and sulfate conjugates,and undergoes enterohepatic circulation[2].
Evaluation of bioequivalence requires pharmacokinetic plotting of time–concentration profle to be accurate.A method for simultaneous extraction is required to extract analyte of interest selectively without co-extracting conjugated metabolites of these drugs which may be back-converted to the parent drug during the derivatization procedure.
A few assays have been reported for individual analysis of ethinyl estradiol and levonorgestrel in human plasma.However, they either lack the sensitivity required especially for ethinylestradiol or do not meet the simultaneous estimation requirements of the proposed work[3–14].Matějíček and Kubáň[5]reported a method for simultaneous analysis;however,the method was not optimized for estimation of ethinyl estradiol and levonorgestrel in human plasma samples.In another report,a semiautomated method for the simultaneous determination of oral contraceptives concentration in human plasma was reported with shorter run time[10].However,the method lacked the required sensitivity level as the lower limit of quanti f cation(LLOQ)for ethinyl estradiol was 10 pg/mL,which was almost 10%of the reportedCmax.
The method presented has the highest extensive range of linearity 4.00–500 pg/mL(125 times)and 0.100–20.0 ng/mL(200 times)compared with the reported methods for ethinyl estradiol and levonorgestrel in human plasma.The plasma volume for sample preparation was 500 μL,which was considerably less than or similar to that in other reported methods[3–4].The on-column loading of ethinyl estradiol and levonorgestrel at LLOQ was only 300 fg and 7.5 pg per sample injection volume respectively,which was signi f cantly lower than that in all other reported procedures [3–14].The proposed method was validated and its application to sample analysis was performed using the Watson LIMS software, which provided excellent data integrity,and they are essential requirements of current regulatory bodies.None of the methods and application were presented with the same.The results and discussion of the incurred sample reanalysis(ISR),which were obtained after implementing the proposed method,have not been discussed or presented in any of the reported methods[3–5].
2.Experimental
2.1.Chemicals and materials
tandards of ethinyl estradiol(Lot No.∶Q0162; 100.0%purity)and levonorgestrel(Lot No.∶F0H323;99.3%purity) were obtained from USP(Rockville,USA).Ethinyl estradiol-D4 (97.1%purity)and levonorgestrel-D6(99.9%purity)as internal standard(IS)were obtained from Clearsynth Labs Limited (Mumbai,India)and TLC Pharmachem Inc.(Concord,Canada), respectively.Dansyl chloride was obtained from Sigma Aldrich (Bengaluru,India).Sodium bicarbonate and sodium hydroxide of GR grade were procured from S.d.Fine Chem Private Limited (Mumbai,India).Formic acid and ammonia solution of GR grade were procured from Merck Private Limited(Mumbai,India).HPLC grade methyl-tert-butyl ether,acetone,acetonitrile and methanol were procured from J.T.Baker Private Limited(Mumbai,India). Water used in the entire analysis was prepared by the Milli-Q water puri f cation system from Millipore(Bengaluru,India).Blank human plasma with sodium heparin as an anticoagulant was obtained from clinical laboratory Supratech Micropath(Ahmedabad, India).Blank plasma was stored at–20°C until use.
2.2.Liquid chromatography and mass spectrometric conditions
A UFLC XR prominence system(Kyoto,Japan)consisting of an LC-20AD XR prominence pump,an SIL-20AC XR prominence autosampler,a CTO-20AC XR prominence column oven and a DGU-20A3 prominence degasser was used for setting the reverse-phase liquid chromatographic conditions.The separation of both analytes and respective internal standard(IS)was performed on a Phenomenex analytical column,type Kinetex C18(50 mm×4.6 mm,2.6 μm). Column temperature was maintained at 30°C in column oven.The mobile phase consisted of 0.1%(v/v)formic acid in water∶acetonitrile with gradient elution from 60%to 90%of acetonitrile composition over a run time of 9.0 min.For gradient elution,the f ow rate of the mobile phase was kept at 0.7 mL/min.The total chromatographic run time was 9.0 min.The autosampler temperature was maintained at 5°C,and the pressure of the system was in the vicinity of 2000 psi.
Ionization and detection of analytes and IS were carried out on a triple quadrupole mass spectrometer,AB SCIEX API-5500(Toronto, Canada),equipped with electrospray ionization(TIS interface of the API 5500)and operated in the positive ion mode.Quantitation was performed using multiple reaction monitoring(MRM)mode to monitor parent→product ion(m/z)transitions 313.3→245.3 for levonorgestrel(Fig.1)and 319.3→251.3 for levonorgestrel-D6 as an IS(f gure not shown).Ethinyl estradiol and ethinyl estradiol-D4 as an IS were quantitatively derivatized with dansyl chloride.The mass transitions for both compounds were 530.1→171.0(Fig.2)and 534.1→171.0(f gure not shown),respectively.The source dependent parameters maintained for all analytes were Gas 1(Nebulizer gas)∶40.0 psig;Gas 2(heater gas f ow)∶60.0 psig;ion spray voltage (ISV)∶5000.0 V,turbo heater temperature(TEM)∶550.0°C;interface heater(Ihe)∶ON;entrance potential(EP)∶10.0 V;collisional activated dissociation(CAD)∶8 psig;and curtain gas(CUR),nitrogen∶30 psig.Compound speci f c values of mass spectrometer parameters are listed in Table 1.Analyst software version 1.6.2 was used to control all parameters of liquid chromatography(LC)and mass spectrometry(MS).WATSON LIMS software version 7.3 was used for regression and f nal data processing.
2.3.Standard stock,calibration standards and quality control sample preparation

Fig.1.Product ion mass spectra of levonorgestrel(m/z313.3→245.3,scan range 100–350 amu)in the positive ionization mode.
The standard stock solutions of ethinyl estradiol(0.1 mg/mL) and levonorgestrel(1 mg/mL)were prepared by dissolving requisite amount of them in methanol.Calibration standards andquality control(QC)samples were prepared by spiking(2%of total plasma volume)blank plasma with serially diluted spiking solutions.Calibration curve standards were made at 4.00,8.00,15.0, 30.0,60.0,120,250 and 500 pg/mL for ethinyl estradiol and 0.100, 0.200,0.500,1.00,2.00,5.00,10.0 and 20.0 ng/mL for levonorgestrel, respectively,while QC samples were prepared at f ve concentration levels,viz.400 pg/mL(high quality control(HQC)),200/48.0 pg/mL (medium quality control(MQC1/2)),12.0 pg/mL(low quality control (LQC))and 4.00 pg/mL(LLOQ QC)for ethinyl estradiol.The QC concentrations of 16.0,8.00,1.50,0.300 and 0.100 ng/mL were applied for levonorgestrel.Stock solutions of ethinyl estradiol-D4 (0.100 mg/mL)and levonorgestrel-D6(1.00 mg/mL)were prepared by dissolving 2.0 mg each of them in appropriate volumes of methanol.Mixed working IS solution containing 5.0 ng/mL ethinyl estradiol-D4 and 80.0 ng/mL levonorgestrel-D6 was prepared by appropriate dilution of the stock solution in methanol.All the solutions(standard stock,calibration standards and quality control samples)were stored at 2–8°C until use.

Fig.2.Product ion mass spectra of dansyl chloride derivatized ethinyl estradiol(m/z530.1→171.0,scan range 100–540 amu)in the positive ionization mode.
Table 1Values of compound speci f c mass spectrometer parameters.
2.4.Sample extraction protocols
Prior to analysis,all frozen subject samples,calibration standards and QC samples were thawed and allowed to equilibrate at room temperature.
To an aliquot of 500 μL of spiked plasma sample,50 μL of mixed IS solution and 500 μL of 2%(v/v)ammonia solution in acetonitrile were added and vortex mixed for 1 min to precipitate the plasma proteins.Extraction of analytes and IS was done in 2.0 mL of methyl-tert-butyl ether∶n-hexane(50∶50,v/v)solvent mixture on a rotary mixer(rotospin)for 10 min at 32g.Samples were centrifuged at 3208gfor 5 min at 10°C.The organic layer(2.0 mL)was separated and evaporated to dryness in a thermostatically controlled water bath maintained at 55°C under a gentle stream of nitrogen.The dried samples were derivatized with 200 μL of sodium bicarbonate solution(pH 11.0,50 mM)and 200 μL of dansyl chloride solution(1 mg/mL)in acetone by incubating the samples for 10 min in water bath maintained at a temperature of 55°C. After incubation completion,the samples were again subjected to liquid–liquid extraction(LLE)to separate out dansylinic acid waste product during the derivatization process.2.5 mL of n-heptane was added to all the samples.The samples were extracted on a rotary mixer for 10 min at 32gand centrifuged at 3208gfor 5 min at 10°C.The organic layer(2.0 mL)was separated and evaporated to dryness in a thermostatically controlled water-bath maintained at 55°C under a gentle stream of nitrogen.The dried samples were reconstituted with 100 μL of mobile phase,vortexed to mix for 10 s,and 15 μL was injected into the chromatographic system.
2.5.Method validation procedures
The bioanalytical method was fully validated following the United States Food and Drug Administration(USFDA)guidelines [15].System suitability experiment was performed by six consecutive injections using the aqueous standard mixture of both analytes and their IS at the start of each batch during method validation.System performance was studied by injecting one extracted blank(without analyte and IS)and one ULOQ(the upper limit of quanti f cation)sample and one LLOQ sample with respective IS at the beginning of each analytical batch and before reinjecting any sample during method validation.Carryover effect of autosampler was checked to verify any carryover of the analyte at the start and at the end of each batch.The design of the experiment comprised the following sequence of injectionsviz.extracted blank sample→ULOQ sample→two extracted blank samples→LLOQ sample.
Selectivity of the method towards endogenous plasma matrix components was assessed in nine different batches of plasma,of which seven were normal sodium heparin plasma and one each of lipidemic and haemolyzed plasma.Selectivity of the method towards commonly used medications in human volunteers was evaluated for acetaminophen,cetirizine,domperidone,ranitidine, diclofenac,ibuprofen,nicotine and caffeine in six different batches of human plasma containing sodium heparin as the anticoagulant.
Linearity of the method was determined by analysis of three linearity curves containing eight non-zero concentrations.Area ratio responses for ethinyl estradiol/ethinyl estradiol-D4 andlevonorgestrel/levonorgestrel-D6 obtained from multiple reaction monitoring were used for regression analysis.Each calibration curve was analyzed individually using least square weighted(1/x2) linear regression which was f nalized during pre-method validation.A correlation coef f cient(r2)value of greater than 0.99 was desirable for all the calibration curves.The lowest standard on the calibration curve was accepted as LLOQ,if the analyte response was at least f ve times more than that of drug-free(blank)extracted plasma.In addition,the analyte peak of the LLOQ sample should be identi f able,discrete and reproducible with a precision (%CV)less than 20%and accuracy within 80%–120%.Deviation of the standards other than LLOQ from nominal concentration should not be more than±15%.
For determining intra-batch accuracy and precision,replicate analyses of plasma samples were performed on the same day.The run consisted of a calibration curve and six replicates of LLOQ QC, LQC,MQC2,MQC1 and HQC samples.Inter-batch accuracy and precision were assessed by analyzing f ve precision and accuracy batches on three consecutive validation days.Precision at each concentration level from the nominal concentration should not be greater than 15%.Similarly,the mean accuracy should be within 85%–115%,except for the LLOQ QC where it should be from 80%to 120%of the nominal concentration.Ion suppression/enhancement effects on the multiple reaction monitoring liquid chromatography-tandem mass spectrometry(MRM LC–MS/MS)sensitivity were evaluated by the post-column analyte infusion experiment. Standard solution containing ethinyl estradiol,levonorgestrel(at ULOQ level)and both IS was infused into the post column via a‘T’connector into the mobile phase at 10 μL/min employing infusion pump.Aliquots of 15 μL of extracted control plasma were then injected into the column by the autosampler.Any dip in the baseline upon injection of double blank plasma(without IS)would indicate ion suppression,while a peak at the retention time of analyte or IS indicated ion enhancement.
Relative recovery(RE),absolute matrix effect(ME)and process ef f ciency(PE)were assessed.All three parameters were evaluated at HQC,MQC1,MQC2 and LQC levels in six replicates.RE was calculated by comparing the mean peak area response of extracted samples(spiked before extraction)with that of unextracted samples(spiked after extraction)at each QC level.Recovery of IS was estimated in the similar way.ME was assessed by comparing the mean peak area response of unextracted samples(spiked after extraction)with mean peak area of neat standard solutions. Overall PE was calculated as(ME×RE)/100%.
All stability results were evaluated by measuring the area ratio response(drug/IS)of stability samples against freshly prepared comparison standards with identical concentration.Stock solutions of analytes and IS were checked for short-term stability at room temperature and long-term stability at 5°C.The solutions were considered stable if the deviation from nominal value was within±10.0%.Autosampler stability(extract stability at 2–8°C and at ambient temperature),bench-top(at room temperature) and freeze–thaw(f ve cycles)stability experiments were performed at LQC and HQC levels using six replicates.Freeze–thaw stability was evaluated by successive cycles of freezing(at-20 and-70°C)and thawing(without warming)at room temperature.Long-term stability of spiked plasma samples stored at-20 and-70°C was also studied at both levels.The samples were considered stable if the deviation from the mean calculated concentration of freshly thawed QC samples was within±15.0%.
To authenticate ruggedness of the proposed method,it was performed with two precision and accuracy batches.The f rst batch was analyzed by different analysts,while the second batch was studied on two different columns.Dilution integrity experiment was evaluated by spiking the QC sample at 800 pg/mL ethinyl estradiol and 32.0 ng/mL levonorgestrel concentration in the screened plasma.The precision and accuracy for dilution integrity standards at 1/10th dilution were determined by analyzing the samples against calibration curve standards.
2.6.Bioequivalence study design and incurred sample reanalysis
Design of the study comprised a randomized,open label,two treatment,two period,two sequence,single dose,crossover bioequivalence study of levonorgestrel/ethinyl estradiol tablets 0.15 mg/0.03 mg(two tablets as a single dose)of test formulation and Microgynon®30 ED(levonorgestrel/ethinyl estradiol tablets 0.15 mg/0.03 mg)tablets(two tablets as a single dose)of Bayer Australia Limited in healthy,adult,female,human subjects under fasting conditions.Each subject was judged to be in good health through medical history,physical examination and routine laboratory tests.Written consent was taken from all the subjects after informing them about the objectives and possible risks involved in the study.An independent ethics committee constituted as per Indian Council of Medical Research(ICMR)approved the study protocol.The study was conducted strictly in accordance with guidelines laid down by the International Conference on Harmonization and USFDA.The subjects were fasted 10 h before administration of the drug formulation.They were orally administered a single dose of test and reference formulations after a recommended wash out period of 28 days with 240 mL of water. Blood samples were collected at the pre-dose,0.33,0.67,1.00,1.25, 1.50,1.75,2.00,2.33,2.67,3.00,3.50,4.00,6.00,8.00,11.00,15.00, 18.00,24.00,36.00,48.00,60.00 and 72.00 h after oral administration of the dose for test and reference formulations in labeled sodium heparin vacutainer.The maximum volume of blood withdrawn during the entire study was not more than 362 mL, which included 322 mL for pharmacokinetic analysis,10 mL for screening,10 mL for post-study safety assessment(hematology and biochemical tests),while a total of 20 mL of heparinized blood was discarded prior to sampling through the venous cannula for each subject in both the periods.Plasma was separated by centrifugation and kept frozen at-20°C until completion of the period and then at-70°C until analysis.During the study,subjects had a standard diet,while water intake was unmonitored.An incurred sample re-analysis(assay reproducibility test)was also conducted by computerized random selection of subject samples. 10%of the total samples were re-analyzed for incurred sample reanalysis experiment.The selection criteria included minimum two samples per period,which were close to the Cmaxand the elimination phase in the pharmacokinetic pro f le of the drugs.The results obtained were compared with the data obtained earlier for the same sample using the same procedure.The percentage change in the value should not be more than±20%.

2.7.Statistical analysis
The pharmacokinetic parameters of ethinyl estradiol and levonorgestrel were estimated by a non-compartmental model using WinNonlin software version 5.2.1(Pharsight Corporation, Sunnyvale,CA,USA).TheCmaxvalues and the time to reach maximum plasma concentration(tmax)were estimated directly from the observed plasma concentration vs.time data.The area under the plasma concentration–time curve from time 0 to 72 h(AUC0–72)was calculated using the linear trapezoidal rule.The AUC0-infwas calculated as∶AUC0-inf=AUC0–72+Ct/Kel,whereCtis the last plasma concentration measured andKelis the elimination rate constant;Kelwas determined using linear regression analysis ofthe logarithm linear part of the plasma concentration–time curve. Thet1/2was calculated as∶t1/2=ln 2/Kel.To determine whether the test and reference formulations were pharmacokinetically equivalent,Cmax,AUC0–72,AUC0-infand their ratios(test/reference) using log transformed data were assessed.Their means and 90% con f dence intervals(CIs)were analyzed using SAS®software version 9.1.3(SAS Institute Inc.,Cary,NC,USA).The drugs were considered pharmacokinetically equivalent if the difference between the compared parameters was statistically non-signi f cant (P≥0.05),and the 90%CIs for these parameters fell within 80%–125%.
3.Results and discussion
3.1.Method development
To develop a selective,rugged and reliable method for the simultaneous estimation of ethinyl estradiol and levonorgestrel in human plasma,the three commonly used extraction procedures were systematically investigated.The chromatographic and mass spectrometric conditions were suitably optimized to get the desired sensitivity,selectivity and linearity in regression curves.
3.1.1.Mass spectrometry
Levonorgestrel and ethinyl estradiol were tuned in atmospheric pressure chemical ionization(APCI)mode considering its nonpolar steroidal moiety.Though levonorgestrel produced a high intense signal in APCI mode and ethinyl estradiol in derivatized or underivatized form did not produce the required signal in APCI mode,the electrospray ionization(ESI)mode with positive polarity was used, which provided a good signal for both analytes.The dansyl derivatization of ethinyl estradiol introduced basic secondary nitrogen into the molecule that was readily ionized in acidic HPLC mobile phases. The derivative showed an intense protonated molecular ion atm/z530.1 under positive turbo ion spray ionization.The collision-induced dissociation of this ion formed a distinctive product ion atm/z171.0, corresponding to the protonated 5-(dimethylamino)naphthalene moiety.The selected reaction monitoring,based on them/z530.1→171.0 transition,was used for ethinyl estradiol.This allowed for an LLOQ of 4.00 pg/mL for ethinyl estradiol and 0.100 ng/mL for levonorgestrel using a 500 μL plasma sample and injecting 15 μL of dansylated derivative into the Shimadzu UFLC XR prominence autosampler coupled to an AB Sciex API 5500 triple quadrupole mass spectrometer.Though dansyl chloride also reacted with amines in the samples,the selected reaction monitoring,based on them/z530.1→171.0 transition,was made speci f c for ethinyl estradiol by separating the analyte of interest peak chromatographically using gradient elution pattern and ultimately eliminating the background noise observed at retention time from blank plasma.Initially,the precursor and product ions were optimized by infusing 200 ng/mL solutions in the mass spectrometer betweenm/z100 and 550.Q1 MS full scan spectra for ethinyl estradiol and its IS predominantly contained protonated precursor[M+H]+ions atm/z530.1 and 534.1 of their dansylated derivatives,respectively.The most abundant and consistent common product ions in Q3 MS spectra for ethinyl estradiol and its IS were observed atm/z171.0 by applying collision energy of 50 eV.The source dependent and compound dependent parameters were suitably optimized to obtain a consistent and adequate response to the analyte.A dwell time of 200 ms for the drug and 50 ms for its IS was adequate and no cross talk was observed between their MRMs.
3.1.2.Optimization of extraction technique
Reported procedures for the estimation of ethinyl estradiol in human plasma have used either LLE or solid phase extraction for sample preparation with little or no information on ion suppression or matrix interference.Considering the steroidal moiety in chemical structures of both analytes and the high log P value, protein precipitation followed by LLE was tried by using the various combinations of organic solvents like diethyl ether,ethyl acetate,methyl tert-butyl ether,n-hexane and n-heptane.The samples were precipitated with the equal amount of acetonitrile to plasma,and LLE with methyl tert-butyl ether combined with n-hexane in a proportion of 50∶50(v/v)gave good response and desired recovery through the extraction.After selective extraction of both analytes,the organic supernatant layer was separated and evaporated to dryness.The dried residue was subjected to dansyl derivatization in alkaline pH at an incubation temperature of 55°C for 10 min.After derivatization,to protect the silica-based column from exposing to high alkaline pH incorporated during the derivatization procedure,the derivatives were re-extracted in organic solvent with n-heptane.To reconstitute the f nal product,various combinations of ammonium acetate,formic acid or ammonium formate solutions with acetonitrile were tried.The samples were reconstituted with initial mobile phase composition as 0.1%(v/v) formic acid in water∶acetonitrile(40∶60,v/v),which provided help to improve the sensitivity,compatibility and reproducible response.
3.1.3.Optimization of chromatographic conditions
To have a rugged and ef f cient chromatography,efforts were made to minimize matrix interference,achieve adequate run time in order to ensure high throughput and attain high sensitivity with good peak shapes.The analytical potential of four different reversedphase columns was evaluated,namely,Atlantis C18(100 mm× 2.1 mm,3 μm),Kinetex C18(50 mm×4.6 mm,2.6 μm),Kinetex PFP (50 mm×4.6 mm,2.6 μm)and Kinetex C18(50 mm×2.1 mm, 2.6 μm)analytical columns.Separation was tried using various combinations of methanol/acetonitrile in acidic buffer(2–20 mM ammonium formate)and additives like formic acid(0.01–0.1%)on these columns to f nd the optimal mobile phase that produced the best sensitivity,ef f ciency and peak shape.Levonorgestrel required a relatively higher portion of aqueous composition to separate closely eluting interferences on selected MRM,while derivatized ethinyl estradiol required a higher portion of organic composition to elute out in relatively shorter retention time while still maintaining the selectivity.The required sensitivity and selectivity at femtogram level on the column were adversely affected by high background noise and many similar MRM transition peaks for ethinyl estradiol.Furthermore,it was required to wash out the retained interferences in the column after elution of the analyte.Hence,careful optimization of chromatography was needed with gradient programming starting from lower organic portion(60%)till separation of levonorgestrel peak to 70%organic portion till separation of ethinyl estradiol and providing nonpolar interference wash out with 90%of organic proportion.In the present work,the best chromatographic conditions as a function of analyte peak intensity,peak shape,adequate retention and analysis run time were achieved with Kinetex C18(50 mm×4.6 mm,2.6 μm)using 0.01%(v/v)formic acid in water∶acetonitrile(40∶60 to 10∶90(v/v)gradient programming)as the mobile phase.The total chromatographic run time was 9.0 min with a retention time of 1.6 and 5.8 min for levonorgestrel and ethinyl estradiol,respectively.The sensitivity achieved for ethinyl estradiol and levonorgestrel was 4.00 pg/mL and 0.100 ng/mL,respectively, which was greater compared with other methods reported in human plasma.Based on the selectivity(unperturbed and stable base line) and signal-to-noise ratio(S/N≥19),it was possible to further lower the LLOQ by about two folds;however,it was not required based on the results of subject samples.Representative MRM ion chromatograms of extracted blank human plasma(double blank)at LLOQ for ethinyl estradiol(Fig.3)and levonorgestrel(Fig.4)demonstrated theselectivity of the method.The chromatograms showed acceptable peak shape for both the drugs.
Ethinyl estradiol-D4 and levonorgestrel-D6 were the deuterated compounds selected as internal standards in the present work.They had similar chromatographic behavior and were easily separated and eluted along with the analytes.There was no effect of IS on analyte recovery,sensitivity or ion suppression.Optimized method was evaluated for interference by the presence of lysophospholipid as shown in Fig.5 and post-column ionization impact as shown in Fig.6.The method was found successfully separating the interferences causing any ionization impact.

Fig.3.MRM ion-chromatograms of(A)double blank plasma(without IS),(B)ethinyl estradiol at LLOQ(m/z530.1→171.0)4.00 pg/mL and IS,and(C)subject sample at 12.6 pg/mL concentration.
3.2.Assay performance and validation
Throughout the method validation,the precision(%CV)of the system suitability test was observed≤3.88%for analyte retention time(RT),IS RT and area ratio of analytes and respective IS,while the signal-to-noise ratio for system performance was≥19.5 and≥92.4 for ethinyl estradiol and levonorgestrel,respectively.Carryover evaluation was performed in each analytical run so as to ensure that it did not affect the accuracy and the precision of the proposed method.No enhancement in the response was observed in the double blank(without analyte and IS)after subsequent injection of the highest calibration standard(aqueous and extracted) at the retention time of the analyte and respective IS.
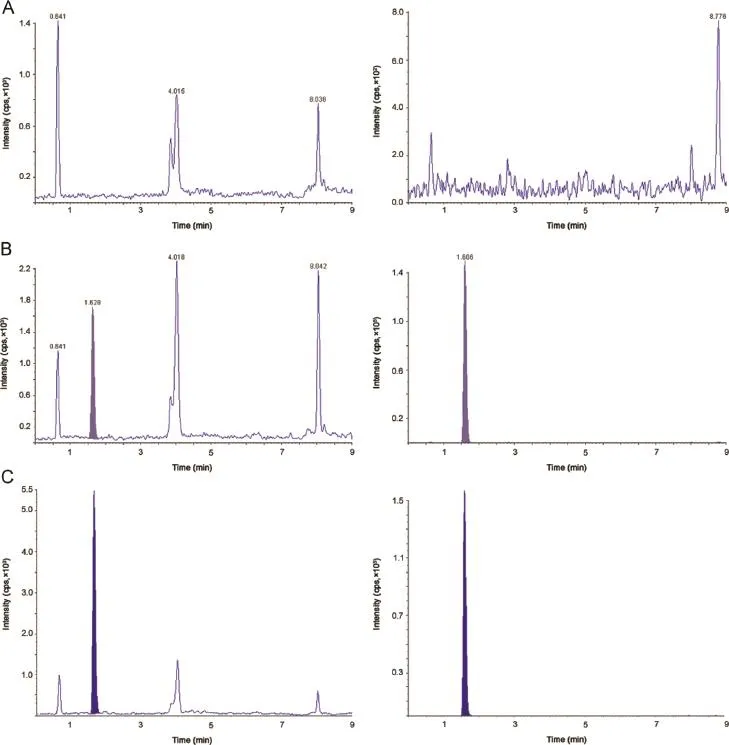
Fig.4.MRM ion-chromatograms of(A)double blank plasma(without IS),(B)levonorgestrel at LLOQ(m/z313.3→245.3)0.100 ng/mL and IS,and(C)subject sample at 0.593 ng/mL concentration.
All three calibration curves were linear over the concentration range of 4.00–500 pg/mL for ethinyl estradiol and 0.100–20.0 ng/mL for levonorgestrel.A straight-line f t was made through the data points by the least square regression analysis, and a constant proportionality was observed.The regression was performed using the Watson LIMS(Laboratory Information Management System)software,version 7.3 to get the high throughput and highest integrity of data without human intervention.The calibration curve(f tted by f rst ordery=ax+b,whereais the slope,bis the intercept,xis the concentration andyis the peak area ratio of drug to IS)was plotted as the peak area ratio(drug to IS)onY-axis vs.the nominal concentration of the drug onX-axis.
The accuracy and precision(%CV)for the calibration curve standards were found within±15.0%for both drugs.The lowest concentration(LLOQ)in the standard curve that could be measured with acceptable accuracy and precision was found to be 4.00 pg/mL for ethinyl estradiol and 0.100 ng/mL for levonorgestrel in plasma at a signal-to-noise ratio(S/N)of≥19.5 and≥92.4, respectively.
The intra-batch and inter-batch precision and accuracy were established from validation runs performed at HQC,MQC1,MQC2, LQC and LLOQ QC levels(Table 2).
The relative recovery and matrix factor data for ethinyl estradiol and IS are presented in Table 3.The relative recovery of the analyte was the‘true recovery’,which was unaffected by the matrix as it was calculated by comparing the peak area ratio response(analyte/IS)of the extracted(spiked before extraction)and unextracted(spiked after extraction)samples.The relative recovery was≥94.06%for ethinyl estradiol and its IS and≥94.78% for levonorgestrel and its IS.Recovery was consistent across all QClevels.Matrix factor was assessed by comparing the mean area response of unextracted samples(spiked after extraction)with mean area of neat standard solutions at four QC levels in six different lots of matrices.CV(%)values for the samples were evaluated and matrix factor was calculated as the mean peak response in the presence of matrix ions divided by mean peak response in the absence of matrix ions.
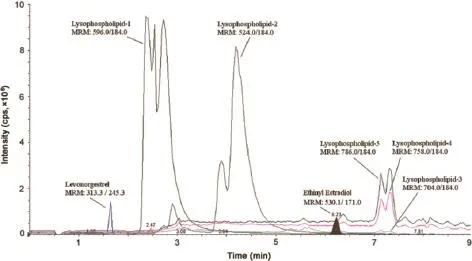
Fig.5.Chromatographic separation of ethinyl estradiol and levonorgestrel from retention time of lysophospholipid.
Fig.6.Injection of three extracted blank plasma samples during post-column infusion experiment of(A)derivatized ethinyl estradiol infusion and(B)levonorgestrel infusion with a chromatogram of the ULOQ sample.
Overall mean IS normalized matrix factor was observed over the range of 0.991–1.019 and 0.994–1.029 for ethinyl estradiol and levonorgestrel,respectively.
The stability of ethinyl estradiol,levonorgestrel and respective IS in human plasma and stock solutions was examined under different storage conditions.Different stability experiments in plasma at two QC levels with the values for percent changes are shown in Table 4.
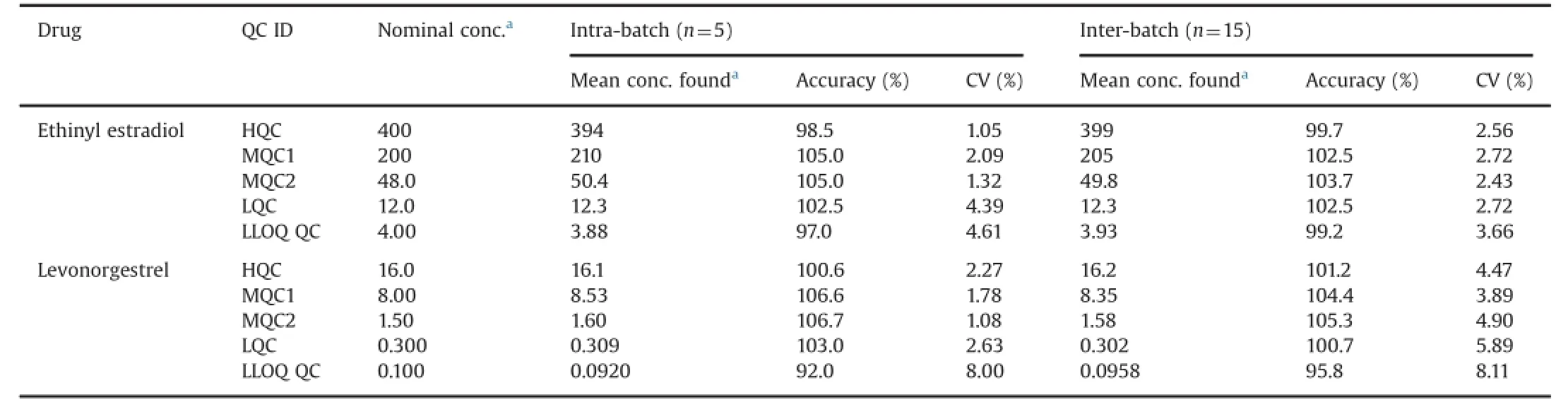
Table 2Intra-batch and inter-batch accuracy and precision.
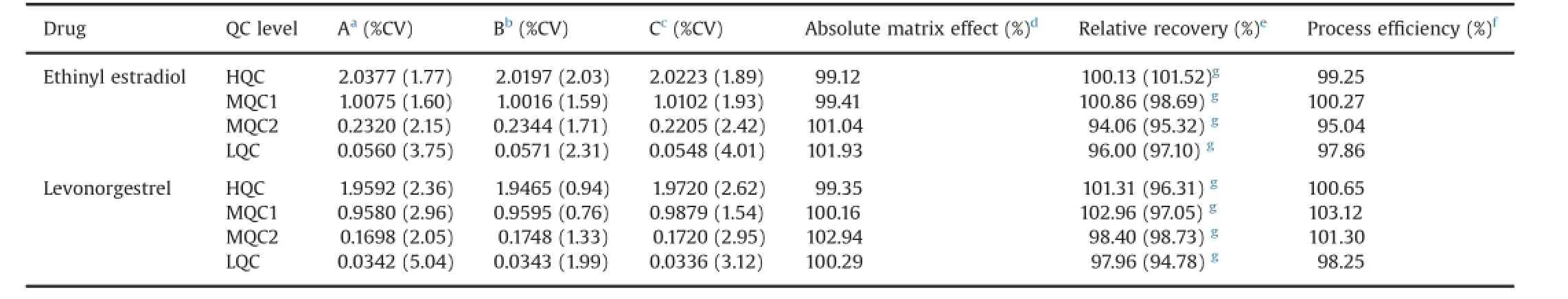
Table 3Absolute matrix effect,relative recovery and process effciency.
Method ruggedness was evaluated using re-injection of the analyzed samples on different columns and mass spectrometer of the same make and with a different analyst.The precision(%CV) and accuracy values for different columns were found≤7.95%and 99.0%–110.0%,respectively,at all four QC levels for ethinyl estradiol and≤1.60%and 101.8%–108.0%for levonorgestrel,respectively.The dilution integrity experiment was performed with the aim to validate the dilution test to be carried out on higher analyte concentration above the ULOQ,which may be encountered during real subject sample analysis.The precision for dilution integrity of 1/10th dilution was 2.59%and 1.00%,while the accuracy results were 107.0%and 110.9%for ethinyl estradiol and levonorgestrel, respectively,which were well within the acceptance limit of 15% for precision(%CV)and 85%–115%for accuracy.
3.3.Application to a bioequivalence study and incurred sample reanalysis
The validated method has been successfully used to quantify ethinyl estradiol and levonorgestrel concentrations in human plasma samples after administration of two tablets dose of test and reference formulations of 0.15 mg/0.03 mg levonorgestrel and ethinyl estradiol,respectively.The study was conducted as per the International Conference on Harmonization,E6 Good Clinical Practice guidelines[16].The method was sensitive enough to monitortheethinylestradiolandlevonorgestrelplasma concentration up to 72 h.In all,approximately 1800 samples including the calibration,QC,volunteer samples and ISR samples were run and analyzed during a period of 40 days,and the precision and accuracy were well within the acceptable limits.The mean pharmacokinetic parameters obtained from the test and reference formulations are presented in Table 5.The 90%CI of the individual ratio geometric mean for test/reference formulation was within 80%–125%for AUC0–72,AUC0-infandCmaxunder fasting conditions as shown in Table 6.Further,there was no adverse event during the study.
Incurred sample reanalysis study has now become an essential part of the bioanalytical process to assess the quality of bioanalytical assays.It reaffrms the reproducibility and reliability of a validated bioanalytical method.This was done by random selection of subject samples(10%of total samples analyzed)[17].Out of 188 incurred samples studied,more than 98%samples showed the change for assay reproducibility within±20%for both the drugs, which authenticated the reproducibility of the proposed method.
4.Conclusions
The bioanalytical methodology for simultaneous determination of ethinyl estradiol and levonorgestrel described in this manuscript can be highly useful for the therapeutic drug monitoring both for analysis of routine samples of single dose or multiple dosepharmacokinetics and for the clinical trial samples and bioequivalence studies with precision,accuracy and high throughput. Date processing was done using the LIMS software which gave the highest data integrity during the method validation and sample analysis.The method involved a sample preparation by protein precipitation,LLE and derivatization followed by LLE.The analytical separation was followed by gradient chromatographic separation in 9.0 min.The validated method was found to be specifc,sensitive,accurate and precise.The established LLOQ was suffciently low to conduct a pharmacokinetic study with 0.03 mg test formulation of ethinyl estradiol and 0.075 mg of levonorgestrel in healthy human volunteers.

Table 4Stability results for ethinyl estradiol and levonorgestrel under different conditions(n=6).
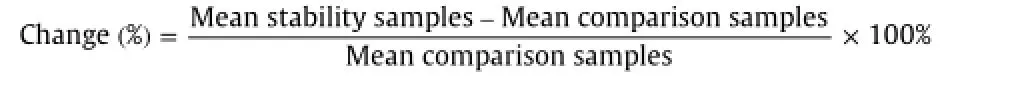

Table 5Mean pharmacokinetic parameters following oral administration of levonorgestrel/ethinyl estradiol[2×(0.15 mg/0.03 mg)]test and reference tablets formulations in 44 healthy Indian subjects under fasting condition.

Table 6Comparison of treatment ratios and 90%CIs of natural log(ln)-transformed parameters for levonorgestrel/ethinyl estradiol[2×(0.15 mg/0.03 mg)]test and reference tablets formulations under fasting condition.
Acknowledgments
The authors are thankful to Director,Mr.Apurva Shah,and chief operating offcer(COO),Dr.Venu Madhav of Veeda ClinicalResearch(India)for providing infrastructure facilities for us carry out this work.The authors are obliged to scientists Mr.Dharmesh Patel and Mr.Gajendra Solanki for their support and quality execution of our research work.
References
[1]Levonorgestrel/Ethinyl Estradiol Information.Available from∶〈http∶//www. drugs.com/cdi/levonorgestrel-ethinyl-estradiol.html〉(accessed 09/02/2014).
[2]Lutera Drug Product Information.Available from∶〈http∶//www.rxlist.com/lu tera-drug/clinical-pharmacology.htm〉(accessed 09/02/2014).
[3]W.Z.Shou,X.Jiang,W.Naidong,Development and validation of a high-sensitivity liquid chromatography/tandem mass spectrometry(LC/MS/MS) method with chemical derivatization for the determination of ethinyl estradiol in human plasma,Biomed.Chromatogr.18(2004)414–421.
[4]J.P.Wheaton,E.E.Chambers,K.J.Fountain,Challenges in developing an ultrasensitive bioanalytical method for ethinylestradiol in human plasma,Bioanalysis 4(2012)769–781.
[5]D.Matějíček,V.Kubáň,High performance liquid chromatography/ion-trap mass spectrometry for separation and simultaneous determination of ethynylestradiol,gestodene,levonorgestrel,cyproterone acetate and desogestrel, Anal.Chim.Acta 588(2007)304–315.
[6]M.R.Anari,R.Bakhtiar,B.Zhu,et al.,Derivatization of ethinylestradiol with dansyl chloride to enhance electrospray ionization∶application in trace analysis of ethinylestradiol in rhesus monkey plasma,Anal.Chem.74(2002)4136–4144.
[7]M.Bonn,U.Eydeler,M.Barkworth,et al.,Bioequivalence study of generic tablet formulations containing ethinylestradiol and chlormadinone acetate in healthy female volunteers,Arzneimittelforschung 59(2009)651–658.
[8]N.C.Borges,R.B.Astigarraga,C.E.Sverdloff,et al.,A novel and sensitive method for ethinyl estradiol quanti f cation in human plasma by high-performance liquid chromatography coupled to atmospheric pressure photoionization(APPI) tandem mass spectrometry∶application to a comparative pharmacokinetics study,J.Chromatogr.B 877(2009)3601–3609.
[9]W.Li,Y.H.Li,A.C.Li,et al.,Simultaneous determination of norethindrone and ethinyl estradiol in human plasma by high performance liquid chromatography with tandem mass spectrometry-experiences on developing a highly selective method using derivatization reagent for enhancing sensitivity,J. Chromatogr.B 825(2005)223–232.
[10]H.Licea-Perez,S.Wang,C.L.Bowen,et al.,A semi-automated 96-well plate method for the simultaneous determination of oral contraceptives concentrations in human plasma using ultra performance liquid chromatography coupled with tandem mass spectrometry,J.Chromatogr.B Anal.Technol. Biomed.Life Sci.852(2007)69–76.
[11]X.F.Liu,C.G.Ding,Q.H.Ge,et al.,Simultaneous determination of gestodene, etonogestrel and ethinylestradiol in plasma by LC–MS/MS following derivatization,Yaoxue Xuebao 45(2010)87–92.
[12]H.B.Theron,C.Coetzee,F.C.W.Sutherland,et al.,Selective and sensitive liquid chromatography–tandem mass spectrometry method for the determination of levonorgestrel in human plasma,J.Chromatogr.B Anal.Technol.Biomed.Life Sci.813(2004)331–336.
[13]Z.Xiong,X.Sun,T.Huo,et al.,Development and validation of UPLC–MS/MS method for simultaneous determination of gestodene and ethinyl estradiol in rat plasma,Biomed.Chromatogr.24(2010)160–168.
[14]L.-Z.Zhao,G.-P.Zhong,H.-C.Bi,et al.,Determination of levonorgestrel in human plasma by liquid chromatography–tandem mass spectrometry method∶application to a bioequivalence study of two formulations in healthy volunteers,Biomed.Chromatogr.22(2008)519–526.
[15]Guidance for Industry∶Bioanalytical Method Validation,U.S.Department of Health and Human Services,Food and Drug Administration,Centre for Drug Evaluation and Research(CDER),Centre for Veterinary Medicine(CVM),2001.
[16]Guidance for Industry∶ICH E6 Good Clinical Practice,U.S.Department of Health and Human Services,Food and Drug Administration,Centre for Drug Evaluation and Research(CDER),Centre for Biologics Evaluation and Research (CBER),1996.
[17]M.Yadav,P.S.Shrivastav,Incurred sample reanalysis(ISR)∶a decisive tool in bioanalytical research,Bioanalysis 3(2011)1007–1024.
Peer review under responsibility of Xi'an Jiaotong University.
*Corresponding author.Tel.∶+91 79 23249069;fax∶+91 79 30013010. E-mail address:prititrivedi.ksv@gmail.com(P.Trivedi).
http∶//dx.doi.org/10.1016/j.jpha.2015.02.002
2095-1779/©2015 Xi'an Jiaotong University.Production and hosting by Elsevier B.V.All rights reserved.This is an open access article under the CC BY-NC-ND license (http∶//creativecommons.org/licenses/by-nc-nd/4.0/).
 Journal of Pharmaceutical Analysis2015年5期
Journal of Pharmaceutical Analysis2015年5期
- Journal of Pharmaceutical Analysis的其它文章
- Application of analytical instruments in pharmaceutical analysis
- JPA Prize in 2014
- Rapid screening and distribution of bioactive compounds in different parts ofBerberis petiolarisusing direct analysis in real time mass spectrometry☆
- Determination of atractylon in rat plasma by a GC–MS method and its application to a pharmacokinetic study☆
- Application of RP–HPLC method in dissolution testing and statistical evaluation by NASSAM for simultaneous estimation of tertiary combined dosages forms☆
- Multiple responses optimization in the development of a headspace gas chromatography method for the determination of residual solvents in pharmaceuticals☆