
Determination of atractylon in rat plasma by a GC–MS method and its application to a pharmacokinetic study☆
2015-01-12 08:02:42HnYnYunyunSunYuyingBinJiXiohongHouZhiguoYuYunliZho
Hn Yn,Yunyun Sun,Yuying M,Bin Ji,Xiohong Hou,Zhiguo Yu,*, Yunli Zho,*
aSchool of Pharmacy,Shenyang Pharmaceutical University,103 Wenhua Road,Shenhe District,Shenyang 110016,China
bSchool of Pharmaceutical Engineering,Shenyang Pharmaceutical University,103 Wenhua Road,Shenhe District,Shenyang 110016,China
Determination of atractylon in rat plasma by a GC–MS method and its application to a pharmacokinetic study☆
Han Yana,Yuanyuan Suna,Yuying Maa,Bin Jia,Xiaohong Houb,Zhiguo Yua,*, Yunli Zhaoa,*
aSchool of Pharmacy,Shenyang Pharmaceutical University,103 Wenhua Road,Shenhe District,Shenyang 110016,China
bSchool of Pharmaceutical Engineering,Shenyang Pharmaceutical University,103 Wenhua Road,Shenhe District,Shenyang 110016,China
A R T I C L E I N F O
Article history:
Received 11 January 2015
Received in revised form
7 March 2015
Accepted 20 March 2015
Available online 28 March 2015Keywords:
Atractylon
Atractylodis
Rat plasma
Pharmacokinetics
GC–MS
A sensitive and selective method based on gas chromatography hyphenated to mass spectrometry(GC–MS)was developed and validated for the determination of atractylon in rat plasma.Plasma samples were processed by liquid–liquid extraction with ethyl acetate-n-hexane(1∶1,v/v)using acetophenone as an internal standard(IS).Analytes were determined in selective ion monitoring(SIM)mode using target ions atm/z108.1 for atractylon andm/z105.1 for acetophenone.The calibration curve was linear over the concentration range of 10–1000 ng/mL with lower limit of quantifcation of 10 ng/mL.The intra-and inter-day precision variations were not more than 10.4%and 9.6%,respectively,whilst accuracy values ranged from-6.5%to 4.9%.Extraction recovery of the assay was satisfactory.This method was successfully applied to quantifcation and pharmacokinetic study of atractylon in rat plasma after intragastric administration of Atractylodis extract.
©2015 Xi'an Jiaotong University.Production and hosting by Elsevier B.V.All rights reserved.This is an open access article under the CC BY-NC-ND license(http∶//creativecommons.org/licenses/by-nc-nd/4.0/).
1.Introduction
Atractylodis(the dried root and stem ofAtractylodes macrocephalaKoidz),commonly known as the‘Baizhu’,is a traditional Chinese medicine(TCM)used for the treatment of lenic asthenia, anorexia,excessive perspiration and abnormal fetal movement[1]. Atractylodis exerts a variety of pharmacological effects such as anti-tumor,anti-infammatory,anti-bacterial and anti-aging effects[2–5].Some studies demonstrated that intragastric administration of a decoction made from Atractylodis has signifcantly increased the immune response induced by a foot-and-mouth disease vaccine in mice[6].In regard to its chemical constituents, the major ingredients of Atractylodis are atractylon(Fig.1), atractylenolides I,II,and III[7].Atractylon is the main active ingredient of Atractylodis[8,9].This compound exhibits various pharmacological activities including anti-infammation,anti-tumor,cytotoxicity against various human cancer cell lines and inhibition of Na+,K+-ATPase activity[10–14].
There are many analytical methods including gas chromatography–mass spectrometry(GC–MS)[15],microbore liquid chromatography(microbore LC)[16]and high performance liquid chromatography(HPLC)[17,18]reported for determining atractylon contained in the crude herb.However,none of these methods have been used for pharmacokinetic studies of atractylon in biological samples.Pharmacokinetic studies of the active ingredients in herbal medicine will contribute substantially to elucidation of their mechanisms of action and development of the quality of herbal medicine.We developed a sensitive and selective GC–MS method for the determination of atractylon in rat plasma using acetophenone(Fig.1)as an internal standard(IS)after liquid–liquid extraction (LLE)with ethyl acetate–n-hexane(1∶1,v/v).The method was fully validated and applied to the pharmacokinetic study after intragastric administration of Atractylodis extract to rats.It was expected that the results of this study would provide some references for the comprehension of the action mechanism and further pharmacological study of atractylon.
2.Materials and methods
2.1.Materials,reagents and animals
The crude herb,Atractylodes macrocephalaKoidz,was purchased from Beijing TongRenTang(Shenyang,China).The raw herbal materials were authenticated by Professor Zhiguo Yu (School of Pharmacy,Shenyang Pharmaceutical University,Shenyang,China).Atractylon(purity≥98%)was purchased from Jianglai Biological Technology Company Limited(Shanghai,China).Acetophenone was used as the IS.Ethanol,n-hexane,methanol, acetonitrile and ethyl acetate(HPLC grade)were purchased from the Concord Tech Reagent Company(Tianjin,China).
Male Sprague-Dawley(SD)rats,weighing 200±20 g,were obtained from Experimental Animal Center of Shenyang Pharmaceutical University(China)and bred with unlimited access to food and water in an air-conditioned animal center at the temperature of 22±2°C and relative humidity of 50%±10% with a natural light–dark cycle.They were fasted with only access to water for 12 h prior to the experiment.Animal experiment was carried out according to the Guideline for Animal Experimentation of Shenyang Pharmaceutical University,and the protocol was approved by the Animal Ethics Committee of the institution.
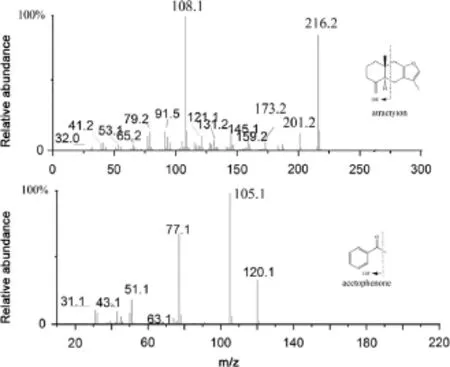
Fig.1.The product ion mass spectra of atractylon and acetophenone.
2.2.Apparatus and operation conditions
2.2.1.Instrumentation and GC–MS conditions
6890N gas chromatography system was f tted with a 5975 mass selective detector of Agilent Technologies(Palo,Alto,CA,USA).GC separation was accomplished on HP-5MS column(30 m×0.25 mm, 0.25 μm),employed with helium as a carrier gas at a f ow rate of 1.0 mL/min.An aliquot(5 μL)of the plasma sample extract was injected in splitless mode.The solvent delay before the MS f lament turned on was set to 2 min to protect the f lament from oxidation. Electron impact(EI)ionization mode was used with nominal electron energy(70 eV).The temperatures of EI ion source,quadrupole mass analyzer,and the interface were maintained at 230,150 and 250°C, respectively.The column temperature was programmed as follows. Initially,the column temperature was maintained at 90°C for 2 min, then increased at a rate of 25°C/min to 150°C,followed by an increase at a rate of 10°C/min to 200°C,and increased at a rate of 25°C/min to 260°C for at least 3 min.The full scan range of the sample for the qualitative analysis was fromm/z30 tom/z500.Fig.1 shows the mass spectra of atractylon and acetophenone.GC–MS was performed in the selective ion monitoring(SIM)mode using target ions atm/z108.1 for atractylon andm/z105.1 for acetophenone(IS).
2.2.2.Preparation of Atractylodis extract
Powdered Atractylodis(200 g)was extracted with 1000 mL of ethanol–water(95∶5,v/v)by re f uxing(1.0 h).Then the extraction solutions were evaporated to dryness using rotary evaporator at a maximum temperature of 50°C and made to a concentration of 5 g of raw herbal materials per milliliter.The content of atractylon in Atractylodis extract was quanti f ed by external standard method using the same chromatography conditions as described above. The content of atractylon in Atractylodis extract was 56.5 mg/mL.
2.2.3.Preparation of stock solutions,calibration standards and quality control samples
Stock solutions of atractylon and IS were prepared in ethanol at a concentration of 1000 and 40 μg/mL.The stock solution of atractylon was diluted with ethanol at a concentration range of 0.2–20 μg/mL,which were used as the working solutions in the linearity experiment.The internal standard solution was obtained by diluting the IS stock solution to a concentration of 4 μg/mL.All solutions were stored at 4°C prior to use.
Calibration standard samples were prepared by spiking 100 μL of blank rat plasma with 5 μL of working solutions.Quality control (QC)samples were also prepared in the same way,using a separately weighed stock solution.The standard and QC samples were extracted on each analysis day with the same procedures for plasma samples as described below.
2.3.Plasma sample preparation
A simple LLE method was applied to extract atractylon and IS from rat plasma.To 100 μL of the rat plasma,5 μL of the IS and 5 μL of ethanol(volume of the corresponding working solution for calibration curve and QC samples)were added.Then the mixture was vortexed for 30 s and extracted with 100 μL of ethyl acetate-nhexane(1∶1,v/v)by shaking on a vortex mixer for 5 min at room temperature.After centrifugation at 13,000 rpm(4°C)for 5 min, 5 μL of the supernatant was injected into the GC–MS for analysis.
2.4.Method validation
The method was validated in terms of speci f city,linearity, precision and accuracy,extraction recovery and stability.
2.4.1.Specifcity
The speci f city of the method was demonstrated by comparing chromatograms obtained from blank plasma samples(from six different drug-free rats),plasma samples spiked with atractylon and the IS,and plasma samples obtained from rats after intragastric administration of Atractylodis extract.
2.4.2.Linearity and the lower limit of quantifcation(LLOQ)
Calibration standard samples were prepared by spiking 100 μL of blank rat plasma with 5 μL of working solutions.The plasma concentration range was 10–1000 ng/mL for atractylon.Calibration curves were created by plotting the peak area ratio of the analyte to internal standard versus concentrations of the analyte in plasma with 1/x2weighted regression.The LLOQ was de f ned as the lowest concentration point of the calibration curve determined with an acceptable deviation from the nominal concentration (±20%).Moreover,signal-to-noise ratio(S/N)of the analyte at this concentration level was at least 10.
2.4.3.Precision and accuracy
QC samples containing atractylon at low,medium and high concentrations of 20,100,and 800 ng/mL were independently prepared for evaluation of precision and accuracy.The intra-and inter-day precisions and accuracies were determined by assaying six replicates of QC samples at three different concentrations on three consecutive days.The variability of determination was depicted as the relative error(RE%)and relative standard deviation (RSD%),respectively.The RE%and RSD%should not exceed 15%.
2.4.4.Extraction recovery
Extraction recovery was determined by comparing the peak areas obtained from analytes in plasma samples which had been spiked with analytes before extraction with those obtained from samples to which analytes had been added after extraction.Extraction recovery was tested at three QC levels in six replicates and should be stable.
2.4.5.Stability
The stability of atractylon was examined by analyzing replicates (n=3)of three levels of QC samples under different conditions. Freeze–thaw stability was evaluated after three cycles of-20 to 20°C.Long-term stability was evaluated by placing plasma samples at-20°C for 7 days.The short-term stability was assessed by analyzing samples kept at room temperature for 5 h.The samples were considered stable if the assay values were within an acceptable deviation from the nominal concentration(±15%).
2.5.Pharmacokinetic study
This validated method was applied to monitor the plasma concentrations of atractylon in rats after intragastric administration of 10 mL/kg of Atractylodis extract(equivalent to 565 mg/kg atractylon).Serial blood samples(approximately 0.3 mL)were collected from fosse orbital vein into 1.5 mL heparinized tubes at 0, 0.17,0.5,1,2,3,4,6,8,12 and 24 h after intragastric administration of Atractylodis extract.Blood samples were immediately centrifuged at 4000 rpm for 10 min.100 μL of supernatant plasma was transferred into another tube and stored at-20°C until analysis. Pharmacokinetic parameters of atractylon were calculated by noncompartmental analysis of plasma concentration versus time data using DAS 2.1 pharmacokinetic program(Chinese Pharmacological Society).
3.Results and discussion
3.1.Method development and optimization
A GC–MS method was developed and optimized for determination of atractylon in rat plasma.An HP-5 capillary column was used for the chromatographic separation.Chromatographic conditions were optimized through several trials to obtain high resolution and relative symmetric peak shapes.Chromatographic conditions like initial(40,90,120,and 150°C)and f nal(220,240, and 260°C)column temperatures,the column temperature rate (5,10,20,and 25°C/min)and the carrier gas f ow rate(0.5,0.8, and 1.0 mL/min)were optimized.The optimized GC separation of analytes was achieved within 15 min,and the retention time of acetophenone and atractylon was 3.8 and 8.9 min,respectively.Acetophenone was found to be optimal as the IS.The SIM mode in the study was selected to avoid the analytical interference of endogenous substance and achieve required selectivity.According to the relative abundance of each fragment ion in mass spectra(Fig.1),analytes were determined using target ions atm/z108.1 for atractylon andm/z105.1 for acetophenone(IS).
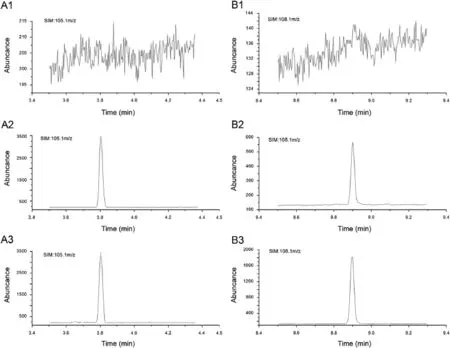
Fig.2.Representative chromatograms of(A)acetophenone and(B)atractylon in(1)blank plasma,(2)blank plasma spiked with atractylon at LLOQ and IS,and(3)plasma samples obtained from rats at 6 h after intragastric administration of Atractylodis extract.
Sample preparation plays an important role in determination of analytes in biological samples.Protein precipitation method was initially attempted with methanol or acetonitrile.However,this technique resulted in low recovery.To obtain clean sample and satisfactory recovery,the LLE method was explored with several organic solvents including diethyl ether,n-hexane,ethyl acetate, ethyl acetate–n-hexane(1∶1,v/v)and ethyl acetate–n-hexane(2∶8, v/v).After several trials,LLE with ethyl acetate–n-hexane(1∶1,v/v) was found to be suitable for determination of atractylon in rat plasma.
3.2.Method validation
3.2.1.Specifcity
Fig.2 represents typical chromatograms of blank plasma,blank plasma spiked with atractylon and the IS,and rat plasma sample collected at 6 h after intragastric administration of Atractylodis extract.Blank rat plasma yielded relative clean chromatograms without interfering peaks both to the analyte and IS.
3.2.2.Linearity and LLOQ
The calibration curves showed good linearity over the concentration range of 10–1000 ng/mL in rat plasma.A typical equation of the calibration curves wasy=3.81×10-3x+2.78×10-2(r=0.9905),whereyis the peak area ratio of atractylon to IS,andxis the concentration of atractylon.The LLOQ for atractylon in rat plasma was 10 ng/mL,which was suffcient for the pharmacokinetic study.
3.2.3.Precision and accuracy
The intra-and inter-day precisions and accuracies of atractylon are summarized in Table 1.The RSD%values were not more than 10.4%and the RE values were all within-6.5%to 4.9%.Results of intra-and inter-day analyses indicated that the proposed GC–MS method was accurate,reliable and reproducible for the quantitative analysis of atractylon in rat plasma samples.
3.2.4.Extraction recovery
Extraction recovery data of atractylon are shown in Table 1. Mean absolute recoveries of atractylon from rat plasma were 91.6%–95.9%at three QC levels,and the extraction effciency of IS was 93.6%±3.4%,which could meet the requirements of analysis. These data indicated that the single-step LLE with ethyl acetate–nhexane(1∶1,v/v)used in the experiment proved to be successful.
3.2.5.Stability
The results of the stability study showed that there was no obvious substance loss under the conditions that plasma samples might experience during this study.The results are summarized in Table 2.

Table 1Precision,accuracy and recovery of atractylon in rat plasma(n=6).

Table 2Stability of atractylon under various storage conditions(n=3).
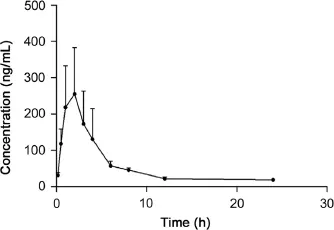
Fig.3.Mean plasma concentration–time curves of atractylon after intragastric administration of Atractylodis extract(mean±SD,n=6).
3.3.Pharmacokinetic study
The validated GC–MS method was successfully applied to the pharmacokinetic study of atractylon in rat plasma following intragastric administration of Atractylodis extract.The mean plasma concentration–time profle of atractylon is shown in Fig.3.The pharmacokinetic parameters including half-time(t1/2),average dwell time(MRT(0–t)and MRT(0–∞)),maximum plasma concentration(Cmax),time to reach the maximum concentrations (tmax),and area under the plasma concentration–time curve (AUC0→tand AUC0→∞)estimated with DAS software using noncompartment model are shown in Table 3.These data showed that low levels of atractylon were seen after a relatively high oral dose.
4.Conclusion
A simple,rapid and sensitive GC–MS method was developed and validated for the quantifcation of atractylon in rat plasma. With no report on GC–MS method for the determination and pharmacokinetic study of atractylon,this is the frst report onpharmacokinetic studies of atractylon in vivo following the intragastric administration of Atractylodis extract.The method presented is valuable for future clinical studies.

Table 3Main pharmacokinetic parameters of atractylon after intragastric administration of Atractylodis extract(n=6).
Acknowledgments
This work was supported by the National Natural Science Foundation of China(Nos.81073143 and 81373926),and Research Fund for the Doctoral Program of Higher Education of China(No. 20092134110004).
[1]Pharmacopoeia of the People's Republic of China,Part I,China Medical Science Press,Beijing,China,2010,95–96.
[2]C.Q.Li,L.C.He,H.Y.Dong,et al.,Screening for the anti-in f ammatory activity of fractions and compounds fromAtractylodes macrocephalakoidz,J.Ethnopharmacol.114(2007)212–217.
[3]Z.Zhang,H.X.Zhang,T.L.Shi,et al.,Research about the antitumor function ofAtractylodes macrocephalakoidz volatile oil,Cancer Res.Clin.18(12)(2006)799–800.
[4]S.I.Jeong,S.Y.Kim,S.J.Kim,et al.,Antibacterial activity of phytochemicals isolated fromAtractylodes japonicaagainst methicillin-resistantStaphylococcus aureus,Molecules 15(2010)7395–7402.
[5]H.J.Li,Z.X.Guo,J.J.Mao,et al.,Anti-aging effect of atractylodisMacrocephalaekoidz decoction on the elderly mice,J.Jiamusi Med.Inst.19(1)(1996)9–10.
[6]F.Xie,Y.T.Li,F.Su,et al.,Adjuvant effect ofAtractylodis macrocephalaeKoidz polysaccharides on the immune response to foot-and-mouth disease vaccine, Carbohydr.Polym.87(2012)1713–1719.
[7]Q.Duan,D.J.Xu,C.X.Liu,et al.,Advances of the rhizome ofAtractylodis macrocephalaeKoidz,Chin.Tradit.Herb.Drugs 39(5)(2008)4–6.
[8]R.B.Zhou,J.Wu,Q.Z.Tong,et al.,Studies on volatile oil fromAtractylodes macrocephalawith different distill methods,J.Chin.Med.Mater.31(2)(2008) 229–232.
[9]J.Wu,R.B.Zhou,Q.Z.Tong,et al.,Optimizing of the parameters in SFE-CO2about volatile oil fromAtractylodes macrocephalaKoidz and studying on its constituents,Mod.Chin.Med.9(4)(2007)14–18.
[10]K.T.Wang,L.G.Chen,L.L.Yang,et al.,Analysis of the sesquiterpenoids in processed Atractylodis Rhizoma,Chem.Pharm.Bull.55(1)(2007)50–56.
[11]C.C.Wang,L.G.Chen,L.L.Yang,et al.,Cytotoxic activity of sesquiterpenoids fromAtractylodes ovataon leukemia cell lines,Planta Med.68 (2002)204–208.
[12]H.K.Kim,Y.K.Yun,Y.J.Ahn,Toxicity of atractylon and atractylenolide III identi f ed inAtractylodes ovatarhizome toDermatophagoides farinaeandDermatophagoides pteronyssinus,J.Agric.Food Chem.55(2007) 6027–6031.
[13]K.Satoh,F.Nagai,K.Ushiyama,et al.,Speci f c inhibition of Na+,K+-ATPase activity by atractylon,a major component of Byaku-jutsu,by interaction with enzyme in the E2 state,Biochem.Pharmacol.51(1996)339–343.
[14]S.Yu,K.Yasukawa,M.Takido,Atractylodis rhizoma extract and its component, atractylon,inhibit tumor promotion in mouse skin two-stage carcinogenesis, Phytomedicine 1(1)(1994)55–58.
[15]F.Q.Guo,L.F.Huang,S.Y.Zhou,Headspace solid-phase microextraction-gas chromatography-mass spectrometry for analysis of volatile components fromAtractylodes macrocephalaKoidz,Chin.J.Chromatogr.25(1)(2007)43–47.
[16]D.Shou,S.W.Dai,J.M.Zhang,et al.,Simultaneous determination of atractylenolide III,atractylenolide I and atractylon inArtactylodis macrocephalausing microbore liquid chromatography,Chin.J.Chromatogr.26(5)(2008)637–639.
[17]K.L.Yan,X.Q.Zhu,L.Zhao,Study on HPLC quantitative quality control and speci f c chromatograms of rhizoma Atractylodis macrocephalae volatile oil, Chin.J.Pharm.Anal.31(4)(2011)758–763.
[18]H.Yin,Z.Q.Wang,L.Wang,et al.,Simultaneous determination of atractylone, Atractylenolide I,II,III inAtractylodes macrocephalaby HPLC-wavelength switching method,J.Tradit.Chin.Med.Pharm.28(1)(2013)233–236.
☆Peer review under responsibility of Xi'an Jiaotong University.
*Corresponding authors.Tel./fax∶+86 24 23986295.
E-mail addresses:zhiguo-yu@163.com(Z.Yu),yunli76@163.com(Y.Zhao).
http∶//dx.doi.org/10.1016/j.jpha.2015.03.002
2095-1779/©2015 Xi'an Jiaotong University.Production and hosting by Elsevier B.V.All rights reserved.This is an open access article under the CC BY-NC-ND license (http∶//creativecommons.org/licenses/by-nc-nd/4.0/).
 Journal of Pharmaceutical Analysis2015年5期
Journal of Pharmaceutical Analysis2015年5期
- Journal of Pharmaceutical Analysis的其它文章
- Application of analytical instruments in pharmaceutical analysis
- JPA Prize in 2014
- High-sensitivity simultaneous liquid chromatography–tandem mass spectrometry assay of ethinyl estradiol and levonorgestrel in human plasma☆
- Rapid screening and distribution of bioactive compounds in different parts ofBerberis petiolarisusing direct analysis in real time mass spectrometry☆
- Application of RP–HPLC method in dissolution testing and statistical evaluation by NASSAM for simultaneous estimation of tertiary combined dosages forms☆
- Multiple responses optimization in the development of a headspace gas chromatography method for the determination of residual solvents in pharmaceuticals☆