
Four new degradation products of doxorubicin∶An application of forced degradation study and hyphenated chromatographic techniques☆
2015-01-12 08:02DheerajKaushikGulshanBansal
Dheeraj Kaushik,Gulshan Bansal
Department of Pharmaceutical Sciences and Drug Research,Punjabi University,Patiala 147002,India
Four new degradation products of doxorubicin∶An application of forced degradation study and hyphenated chromatographic techniques☆
Dheeraj Kaushik,Gulshan Bansal*
Department of Pharmaceutical Sciences and Drug Research,Punjabi University,Patiala 147002,India
A R T I C L E I N F O
Article history:
Received 10 March 2015
Received in revised form
13 May 2015
Accepted 15 May 2015
Available online 23 May 2015Keywords:
Doxorubicin
TOF
Forced degradation
Liquid chromatography
Degradation product
Mass fragmentation pattern
Forced degradation study on doxorubicin(DOX)was carried out under hydrolytic condition in acidic, alkaline and neutral media at varied temperatures,as well as under peroxide,thermal and photolytic conditions in accordance with International Conference on Harmonization(ICH)guidelines Q1(R2).It was found extremely unstable to alkaline hydrolysis even at room temperature,unstable to acid hydrolysis at 80°C,and to oxidation at room temperature.It degraded to four products(O-I–O-IV)in oxidative condition,and to single product(A-I)in acid hydrolytic condition.These products were resolved on a C8(150 mm×4.6 mm,5 μm)column with isocratic elution using mobile phase consisting of HCOONH4(10 mM,pH 2.5),acetonitrile and methanol(65∶15∶20,v/ v/ v).Liquid chromatography–photodiode array(LC–PDA)technique was used to ascertain the purity of the products noted in LC–UV chromatogram.For their characterization,a six stage mass fragmentation(MS6)pattern of DOX was outlined through mass spectral studies in positive mode of electrospray ionization(+ESI)as well as through accurate mass spectral data of DOX and the products generated through liquid chromatography–time of fight mass spectrometry(LC–MS–TOF)on degraded drug solutions.Based on it,O-I–O-IV were characterized as 3-hydroxy-9-desacetyldoxorubicin-9-hydroperoxide,1-hydroxy-9-desacetyldoxorubicin-9-hydroperoxide,9-desacetyldoxorubicin-9-hydroperoxide and 9-desacetyldoxorubicin,respectively,whereas A-I was characterized as deglucosaminyl doxorubicin.While A-I was found to be a pharmacopoeial impurity,all oxidative products were found to be new degradation impurities.The mechanisms and pathways of degradation of doxorubicin were outlined and discussed.
©2015 Xi'an Jiaotong University.Production and hosting by Elsevier B.V.All rights reserved.This is an open access article under the CC BY-NC-ND license(http∶//creativecommons.org/licenses/by-nc-nd/4.0/).
1.Introduction
Drug regulatory agencies such as United States Food and Drug Administration(USFDA)and ICH have laid down very stringent guidelines for the control of impurities in drug substances and products[1–6].ICH Q3A(R2)and Q3B(R2)guidelines specifcally require the identifcation of impurities(process or degradation related)in any drug substance and product[3,4].Identifcation of degradation related impurities(degradation products)remains a major challenge during product development.These arise due to chemical susceptibility of a drug molecule to varied chemical environments during product development,transportation and shelf life.Moreover,these are formed generally in minute amounts, which may not be suffcient to facilitate their characterization. Hence,ICH Q1A(R2)guidelines recommend forced degradation study(stress testing)on drug substance under different chemical environments to facilitate isolation and/or characterization of all possible/major degradation products of the drug[2].
Anthracyclines constitute an important class of anti-tumor chemotherapeutic drugs with a broad spectrum of anticancer activity.DOX(also known as adriamycin)is a naturally anthracycline derivative and is a hydroxylated analog of daunorubicin(Fig.1).It is particularly used for the treatment of disseminated neoplastic conditions such as acute lymphoblastic leukemia,acute myeloblastic leukemia,Wilm's tumor,neublastoma,soft tissue and bone sarcomas,breast carcinoma,ovarian carcinoma,transitional cell bladder carcinoma,thyroid carcinoma,Hodgkin's and non-Hodgkin's lymphomas,bronchogenic carcinoma and gastric carcinoma [7,8].DOX is an important component of multi-chemotherapeutic drug regimen,and is usually given combined with cyclophosphamide,vincristine,bleomycin or prednisone.It is offcial in BritishPharmacopoeia,UnitedStatesPharmacopoeia,Indian pharmacopoeia and Martindale[9–12].The monograph about DOX in British Pharmacopoeia lists four impurities(Imp A–Imp D)[9] (Fig.1).Some studies have reported on degradation and stability of DOX under varied conditions such as aqueous,photolytic and biological environments[13–20].Of these,only Cielecka-Pionteket al.[13]have proposed that on the basis of limited data,DOX and daunorubicin in solid state degrade to apolar degradation products that behave like 7-deoxyaglycons.None of other studies has characterized the degradation products formed under different conditions.Herein,we report(i)a comprehensive forced degradation study on DOX under hydrolytic,photolytic,oxidative and thermal degradation conditions as prescribed in ICH Q1(R2) guidelines;(ii)structural characterization of degradation products through mass fragmentation and accurate mass spectral analyses; and(iii)most plausible mechanisms of DOX degradation.
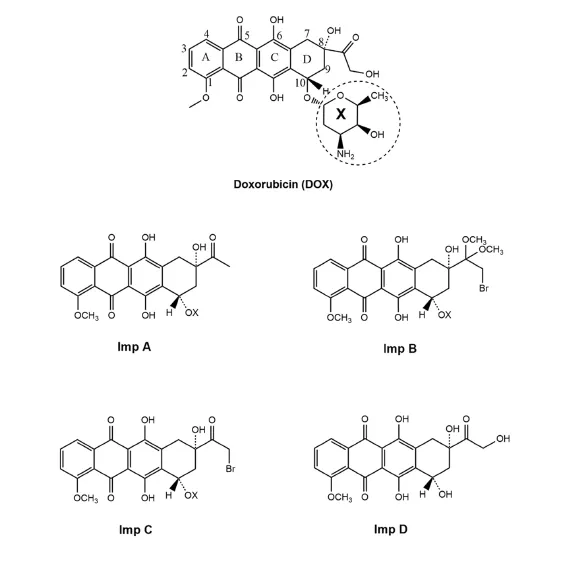
Fig.1.Structure of doxorubicin(DOX)and its known impurities.
2.Experimental
2.1.Drug and chemicals
Doxorubicin hydrochloride(DOX)was generously provided by Strides Arcolabs Pvt.Ltd.(Bangalore,India)as a gift sample.All analytical grade chemicals(sodium hydroxide,hydrochloric acid, hydrogen peroxide(30%)and ammonium formate)were supplied by Loba Chemical Pvt.Ltd.(Mumbai,India).The high performance liquid chromatography(HPLC)grade solvents and chemicals (methanol,acetonitrile and formic acid)were procured from Merck Specialist Pvt.Ltd.(Mumbai,India).HPLC grade water was obtained from Direct Ultra water puri f cation system(Bio-Age Equipment and Services,SAS Nagar,India)in the laboratory.
2.2.Equipments
A high precision water bath and a hot air oven digitally controlling temperature variation of 1 and 2°C,respectively(Narang Scienti f c Works,New Delhi,India)were employed for hydrolytic and thermal degradation studies.Photostability chamber which is equipped with digital controller capable of maintaining temperature and relative humidity(RH)within±2°C and±5%,respectively(KBF 240,WTB Binder,Tuttlingen,Germany)was used for photodegradation studies.The light sources f xed in the chamber provided an illumination bank in compliance with ICH guideline Q1B[21].Liquid chromatography–ultraviolet(LC–UV)analyses of the forced degradation samples were carried out on a a binary HPLC system(515 pumps)equipped with a Rheodyne manual injector and a 2487 dual wavelength detector controlled by Empower 2 software(Waters,Milford,MA,USA).An Agilent C8(150 mm×4.6 mm,5 μm)column was used for chromatographic separation of the drug and degradation products.Purity of peaks of DOX and products in the degradation samples were ascertained by LC–PDA analysis using a binary HPLC system consisting of a 2707 auto-injector and a 2998 PDA detector(Waters,Milford,MA,USA). LTQ-XL ion trap quadrupole mass spectrometer(Thermo Scienti f c, Germany)was used to record multi-stage mass spectral(MSn)data of DOX in positive mode of electrospray ionization(+ESI).For LC–MS–TOF analyses of DOX and degradation products,an Agilent 1100 series LC system(Agilent Technologies Inc.,CA,USA)controlled by Hystar(Ver.3.1)software coupled with a microTOF-Q11 mass spectrometer(Bruker Daltonics GmbH,Germany)controlledby microTOF control software ver.2.0 was used.A splitter allowing entry of only 35%of the eluent was placed before the mass detector in order to avoid signal saturation and detector contamination.
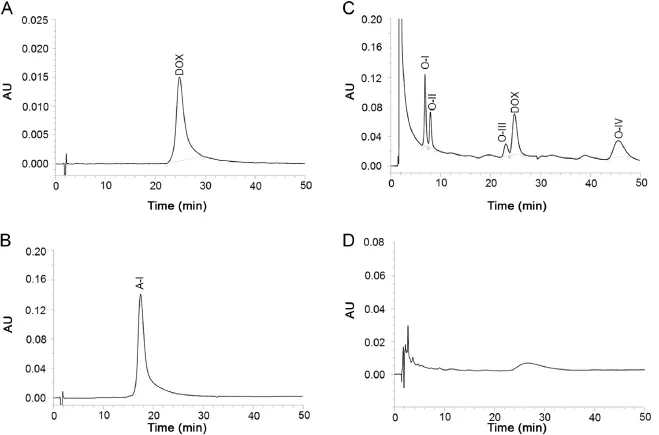
Fig.2.Chromatogram of standard solution of DOX(A),and DOX solutions exposed to 0.1 M HCl at 80°C(B),30%H2O2at room temperature(C),and 0.1 M NaOH at 80°C(D).
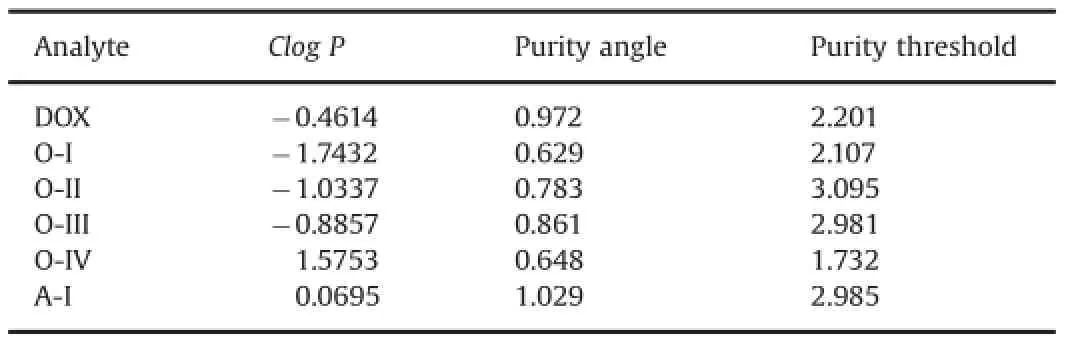
Table 1Clog Pvalues and peak purity data of DOX and its degradation products.
2.3.Forced degradation
DOX was exposed to conditions of hydrolysis,oxidation,dry heat and photolysis.Concentration of DOX in solution/suspension for the study was 0.1%(m/v).Solutions/suspensions of DOX in water,NaOH(0.1 M)and HCl(0.1 M)were exposed to 80°C for a period of 8 h for hydrolytic degradation.Suspension of DOX in H2O2(30%)was placed at 30±5°C in dark for 24 h for oxidative degradation.DOX in solid state was sealed in amber color vials and placed in hot air oven maintained at 50°C for 30 days for thermal degradation.For photolytic degradation,2 mL of a solution of DOX in acetonitrile was mixed with 3 mL of each stressor separately(i.e.0.1 M HCl,0.1 M NaOH and water)in transparent glass vials.These vials as well as the solid drug spreading as a thin layer in a petri dish were placed in the photostability chamber for 14 days to expose the samples to about 200 W h/m2of UV light and 1.2 million lx h of white light.Another similar set of solid drug and drug solutions wrapped well in aluminum foil was also placed in the photostability chamber for the same period of time and served as dark control.Samples from each degradation condition were stored at 4°C till analysis.
2.4.LC–UV and LC–PDA analyses
For chromatographic analysis,the thermal and photodegraded DOX in solid state was solubilized in methanol to produce solutions of concentration 0.1%(m/v).Each of these solutions as well as drug solutions/suspensions exposed to hydrolytic,oxidative and photolytic degradation conditions was diluted(1 in 10)with mobile phase.The drug suspension,wherever formed,was stirred well and sonicated for 5 min to make a homogenous suspension, whereas the acid and alkali degraded drug solutions were neutralized before dilution.Each diluted sample was f ltered through a nylon membrane(0.45 μm,13 mm)before injected on the HPLC system.Blank solution corresponding to each sample was also processed similar to the sample and its LC–UV analysis precedes that of the sample.Chromatographic separation of DOX and its degradation products was achieved by running 20 μL of degraded drug solution on a C8column(150 mm×4.6 mm,5 μm,Agilent) by isocratic elution with mobile phase composed of ammonium formate(10 mM,pH 2.5),acetonitrile and methanol(65∶15∶20,v/ v/ v)at a f ow rate of 1 mL/min at ambient temperature(27±2°C). The eluent was detected at 234 nm.The purity of DOX and all its degradation products peaks in LC–UV chromatograms were established through LC–PDA analysis.
2.5.+ESI-MSnand LC–MS–TOF studies
Six stage mass spectra of DOX in+ESI mode were recorded (+ESI-MS6)using ionization potentials increasing from 18.0 V to 28.0 V,and rationally selecting the precursor ion for each stage. Operating parameters of mass detector such as end plate offsetvoltage,capillary voltage,collision cell RF,nebulizer,dry gas f ow, and dry temperature for recording MS6spectra of DOX as well as LC–MS–TOF spectra of DOX and degradation products were optimized as-500 V,4500 V,400.0 vpp,1.2 bar,6.0 L/min and 200°C, respectively.The LC–MS–TOF analyses were carried out by using ionization potentials of 10 and 15 V.All MS and TOF spectra were recorded over a mass range of 50–1000.Masses of various ion peaks in TOF spectra were recorded up to the fourth decimal precision for accurate mass measurements.

Fig.3.Six stage mass fragmentation spectra of DOX.The precursor ion targeted to record each mass spectrum is marked by“*”.

Table 2Precursor/parent ions and product ions in MS6studies.
3.Results and discussion
3.1.Forced degradation behavior
DOX was detected as a sharp peak at 25.15 min wherein no impurity was noted in LC–UV chromatogram of standard solution (Fig.2A).It degraded to a single product(A-I)under acid hydrolytic condition after 8 h(Fig.2B),and four products(O-I–O-IV) under oxidative condition over 24 h(Fig.2C).LC–PDA studies on acid hydrolyzed and oxidized drug solutions showed that purity angles of the peaks of DOX,O-I,O-II,O-III and O-IV and A-I were less than their purity threshold(Table 1),which indicated that all peaks were pure and no other products co-eluted with any of these peaks.Exposure of the drug to hydrolysis in water at 80°C for 8 h,and to thermal degradation condition for 30 days produced no degradation products.In contrast,its exposure to 0.1 M NaOH at 80°C for 8 h caused its almost total degradation(Fig.2D).Even in mild alkaline medium(0.01 M NaOH)at 40°C(Supplementary Fig.S2A)as well as at room temperature within 30 min,the drug degraded almost completely to several degradation products.In all alkali degradation conditions,the products were eluted ascomplex bunch of peaks within 10–15 min that could not be resolved even after numerous modi f cations under chromatographic conditions such as composition of mobile phase,pH of buffer and stationary phase.Four LC–UV chromatograms showing the best, but still unacceptable resolution are given in Supplementary Fig. S1.The degradation behavior of DOX under acidic and alkaline hydrolytic,and oxidative conditions was in consonant with the f ndings by other laboratories[13–18].Comparison of the LC–UV chromatograms of the drug exposed to photolytic conditions (Supplementary Fig.S2B and D)with those of the drug stored indark(Supplementary Fig.S2C and E)in acidic and alkaline media, respectively revealed that no peak was formed due to photodegradation.Also,no additional photodegradation product was formed when solid drug was exposed to the photolytic conditions (Supplementary Fig.S2F).It indicates that degradation pattern of DOX under photolytic conditions in acidic and alkaline media is similar to that under hydrolytic condition,and hence the drug is suggested to be photostable.

Fig.4.Mass fragmentation pattern of DOX and its acid degradation product(A-I).The dotted arrows outline the fragmentation pathways of A-I.
However,these results of photostability of DOX are found con f icting,at the outset,with those in available reports on photodegradation of DOX.Wood et al.[15,16]have conducted a photodegradation study on DOX at different pH and in different containers.They found that(i)DOX degraded due to cleavage of glycosidic linkage in different media under photolytic conditions;(ii) both the glass and polyethylene container adsorb DOX;and(iii) the rate of drug degradation was inversely proportional to its concentration(i.e.10%degradation in 100 μg/mL solution of DOX and>60%degradation in 10 μg/mL solution after 168 h exposure to visible light).Nonetheless,the f rst two interpretations may not be considered really true with reference to our explanations that are as follows∶Firstly,the glycosidic linkage is known to cleave under mild acid hydrolytic as well as strong alkaline hydrolytic conditions.However,there is no documented proof of its photolytic cleavage.Therefore,DOX might have degraded due to hydrolysis but not photolysis.Secondly,the major decrease in content of DOX in the degraded solution may also be,most likely,due to adsorption of DOX on the sample containers in addition to the actual degradation.The third f nding actually acts as a support to photostability of DOX in the present study because herein we have subjected DOX to different degradation conditions using a solution of concentration 1000 μg/mL,which,according to Wood et al. [15,16]degrades at the least rate.In another study,DOX was found to degrade to 3-methoxysalicyclic acid and an unstable dihydroquinone after exposure to laser beam(λ 483 nm)for 48 h. These two products react with oxygen available in reaction mixture and form DOX and H2O2.Hence,there was no net loss/degradation of DOX[15].Other photodegradation studies have revealed that DOX degrades after exposure to long UV light in the presence of photosensitizers such as ribo f avin.This ribo f avin mediated photooxidation of DOX is further increased in the presence of histidine and urocanic acid[17–19].The results of these literature reports suggest that the photodegradation of DOX is either reversible or is mediated by a photosensitizer.In th present study,the degradation of DOX was carried out using a drug concentration of 1000 μg/mL without any photosensitizer.Hence,the drug does not degrade,and is termed as photostable under the tested conditions.However,in general,as light exposure tends to accelerate most chemical reactions,the drug may be recommended to be stored in containers affording protection from light.In addition,the drug is found stable at neutral pH,but its stability decreases signi f cantly with increasing or decreasing the pH beyond 7.
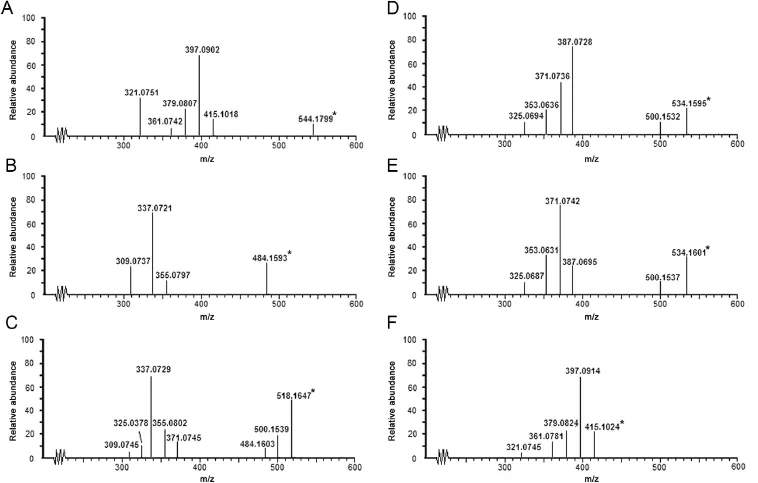
Fig.5.LC–MS–TOF spectra of DOX and its degradation products(*indicates the parent ion).
3.2.Mass fragmentation pattern of DOX
Sleno et al.[22]have reported the mass fragmentation pattern of DOX on the basis of MS3spectra.In the present study,six stage mass fragmentation spectra(MS6)of DOX were recorded(Fig.3) for assisting the characterization of degradation products.The drug was detected atm/z544 as parent ion(M1)in MS1(Fig.3A) corresponding to its molecular mass of 543 Da.The various product ions formed from M1and different precursor ions in all six stages are summarized in Table 2.Based on the comparison of product ions formed from different precursor ions in all mass spectra,a systematic mass fragmentation pattern of DOX was outlined(Fig.4).The product ions formed directly from therespective precursor ion at each stage are marked as bold(Table 2) and subsequently,M1fragmented directly to product ions ofm/z526,415 and 397(Fig.3B)by the loss of water and amino sugar moiety as speci f ed in Fig.4.The heaviest fragmentm/z526(M2a) was used as precursor ion to record MS3aspectrum wherein it fragmented to product ions ofm/z508 by the loss of water andm/z397 by the loss of amino sugar moiety(Fig.3C).The fragment ofm/z415(M2b)fragmented tom/z414 and 397 in MS3bthrough the loss of a hydrogen radical and amino sugar moiety,respectively (Fig.3D).Incidentally,product ion ofm/z397 was formed in MS2, MS3aas well as MS3b.Hence,it was taken as precursor ion to record subsequent mass spectra.Fragmentation ofm/z397(M3) produced product ions ofm/z379(M4)and 337 in MS4spectrum due to the loss of water molecule and a C2H4O2respectively from side chain of ring A(Fig.3E).The heaviest fragment ofm/z379(M4)in MS4spectrum was further fragmented to product ions ofm/z361(M5),321 and 309(Fig.3F).Finally,M5was employed as precursor ion to generate MS6spectrum wherein it fragmented to product ions ofm/z333 by the loss of CO moiety[22]andm/z346 by the loss of a methyl radical(Fig.3G).In addition to MS6study, DOX was also analyzed through LC–MS–TOF to generate accurate mass spectral data.The TOF spectrum of DOX(Fig.5A)showed parent ion atm/z544.1799.The major fragments were noted atm/z526.1619,415.1024,397.0902.379.0807,361.0742 and 321.0751. These masses were found to be very close to the theoretical accurate masses and the structures of product ion ofm/z526,415, 397,379,361 and 321(Table 3)formed during MS6study which supported the proposed structural assignment to these ions.
3.3.LC–MS–TOF studies for characterization of degradation products
Structures of the degradation products were characterized with the help of MS6fragmentation pattern of DOX,comparative accurate mass spectral data of the drug and products generated through LC–MS–TOF analysis of degraded drug solutions,and Elemental Composition Calculator Software that provided the most probable molecular formula corresponding to ion peaks in the mass spectra.The four oxidative products(O-I–O-IV)and the single acid hydrolyzed product(A-I)detected in LC–UV chromatogram were detected in total ion chromatogram(TIC).The LC–MS–TOF spectra of DOX and each product are given in Fig.5 and their accurate mass spectral data are summarized in Table 3 and 4.
3.3.1.Product O-IV
The heaviest peak in MS–TOF spectrum of O-IV was noted atm/z484.1593(Fig.5B)which was assigned as its parent ion(MIV) peak.An even molecular mass of MIVsuggested an odd number nitrogen atom[23].Hence,the single nitrogen atom in amino sugar component of DOX was intact in O-IV.The mass difference of 60.0226 Da between M1and MIVwas found to correspond to molecular formula C2H4O2(60.0231 Da)[24].The possible molecular structures corresponding to C2H4O2include acetic acid, 2-hydroxyacetaldehyde(OHCH2CHO)and 1,2-ethylenediol.Out of these,2-hydroxyacetaldehyde was possible to form from hydroxylacetyl side chain on ring D of DOX.Hence,O-IV was proposed to form by the loss of COCH2OH group from DOX that was possible through Baeyer Villiger oxidation in the presence of H2O2.Based on this proposition,O-IV was suggested to be9-desacetyldoxorubicin,which can exist in two forms due to ketoenol tautomerism.This proposition was supported by a similar kind of product under oxidative conditions reported to form from idarubicin[25].The TOF spectrum of O-IV showed product ions atm/z355.0797,337.0721 and 309.0737.The fragment ofm/z355.0797 was 129.0796 Da less than MIVand was proposed to form by the loss of amino sugar moiety from MIVsimilarly as M2bforming from M1and M2a,respectively.Them/zvalues 337.0721 and 309.0737 matched very closely with accurate theoretical mass of structures assigned to fragmentsm/z337 and 309,respectively, in MS4and MS5spectra of DOX.Based on this comparative mass spectral data,the suggested structure of O-IV was proposed to undergo fragmentation as shown in Fig.6.The fragment ofm/z309.0737 was possible to form fromm/z337.0721 by the loss of CO group(27.9984 Da)similar to mass fragmentation of phenols or alcohols reported in standard literature[24].
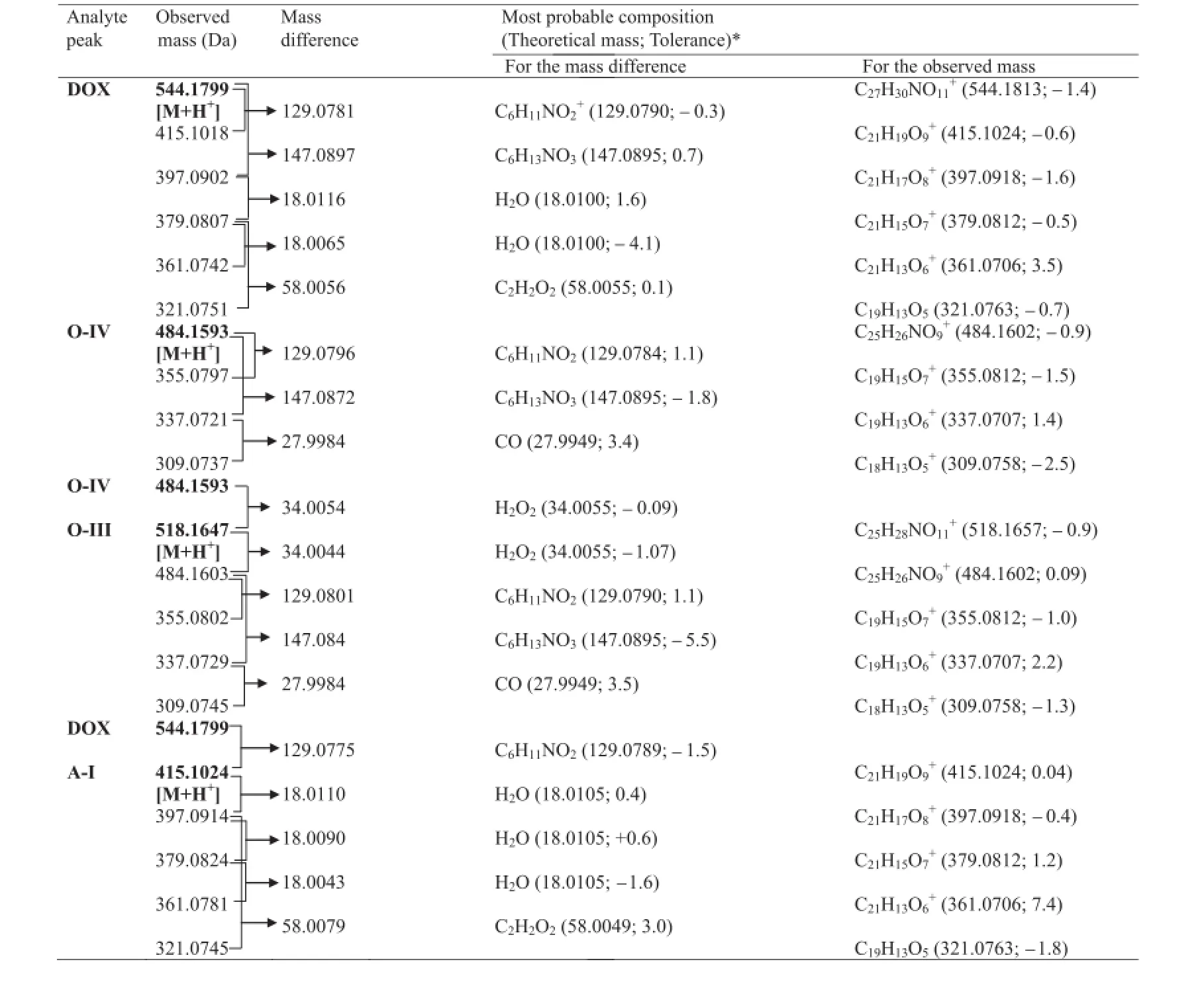
Table 3Molecular formulae corresponding to the measured mass of various peaks in MS–TOF spectra of DOX and products O-IV,O-III and A-I.
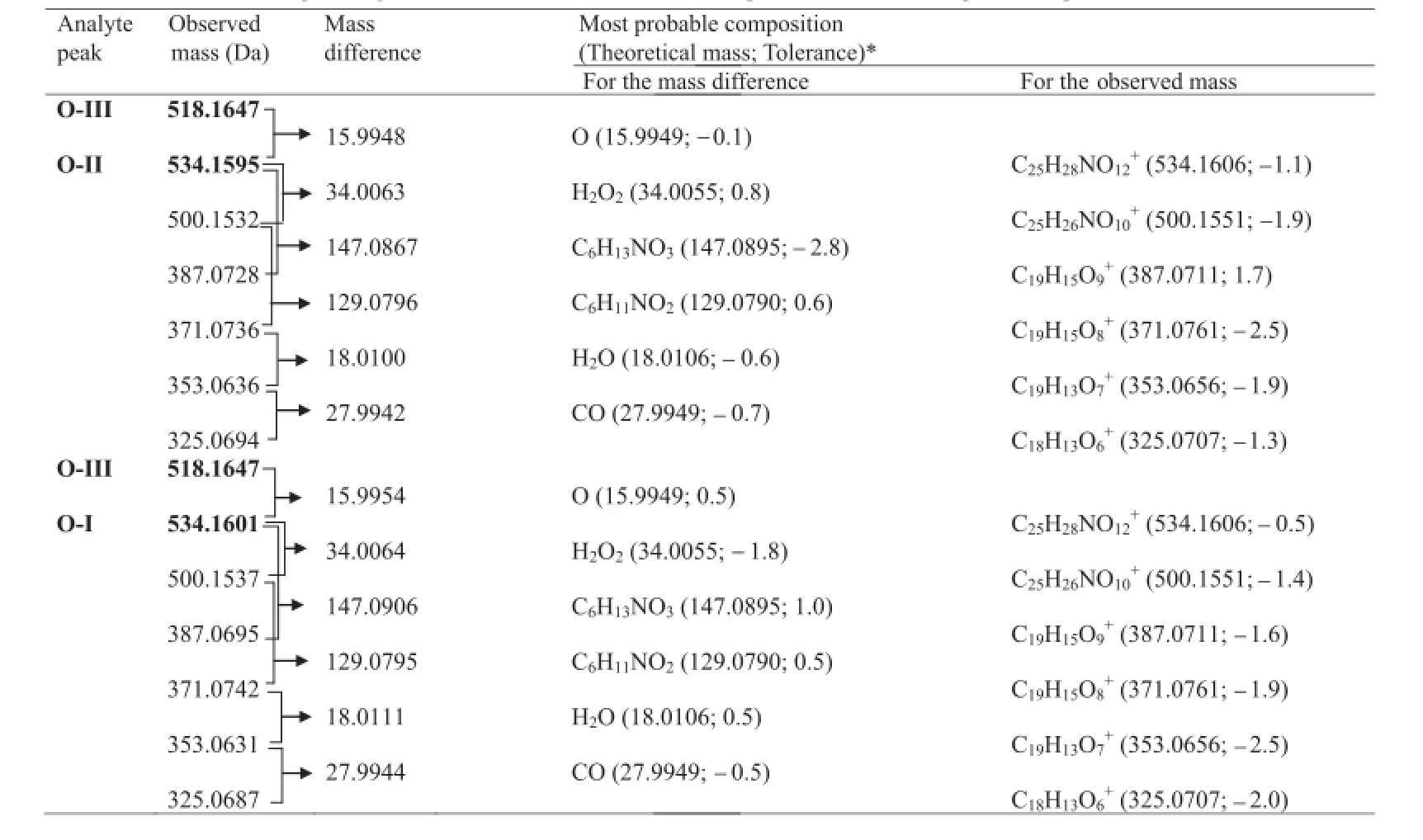
Table 4Molecular formulae corresponding to the measured mass of various peaks in MS–QTOF spectra of products O-I and O-II.
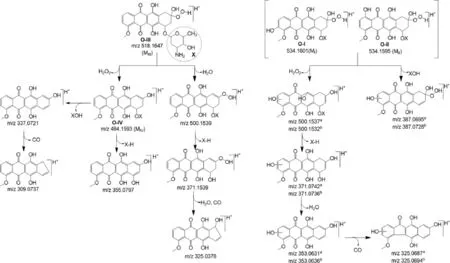
Fig.6.Proposed mass fragmentation pattern of oxidative products(O-I–O-IV).The fragments superscripted with“a”belong to O-I while those with“b”belong to O-II.
3.3.2.Product O-III
It was detected as the heaviest and major peak atm/z518.1647 (Fig.5C)that was assigned as its parent ion(MIII)peak.An even molecular mass of MIIIsuggested an odd number of nitrogen atoms[23].Hence,the single nitrogen atom present in amino sugar component of DOX was suggested to be intact in O-III,similarly as in O-IV.The MS–TOF spectrum also showed fragment ion ofm/z500.1539,484.1603,371.0745,355.0802,337.0729, 325.0378 and 309.0745.Of these,the fragments ofm/z484.1603, 355.0802,337.0729 and 309.0745 were also noted in TOF spectrum of O-IV.It indicated that O-III may be a derivative of O-IV itself.MIIIwas 34.0044 Da heavier thanm/z484.1603,and this mass difference was found to correspond to an H2O2molecule(34.0055 Da) [24].Based on these results,it was suggested that O-III might be formed by the addition of H2O2across the ketone group generated on ring D in keto form of O-IV.This contention was supported by an earlier study,in which a similar hydroperoxide degradation product was proposed to form from idarubicin[25].Hence,O-III was proposed to be 9-desacetyldoxorubicin-9-hydroperoxide, which underwent mass fragmentation(Fig.6)in consonant with its MS–TOF spectrum.The fragment ofm/z484.1603 was possible to form by the loss of H2O2from MIII,whereas the product ions ofm/z355.0802,337.0729 and 309.0745 were proposed to form similarly as from MIV.The product ions ofm/z500.1539 and 371.0745 were possible to form by the loss of H2O molecule followed by the loss of X-H.The fragment ofm/z325.0387 was proposed to form fromm/z371.0745 through one or two steps.
3.3.3.Products O-II and O-I
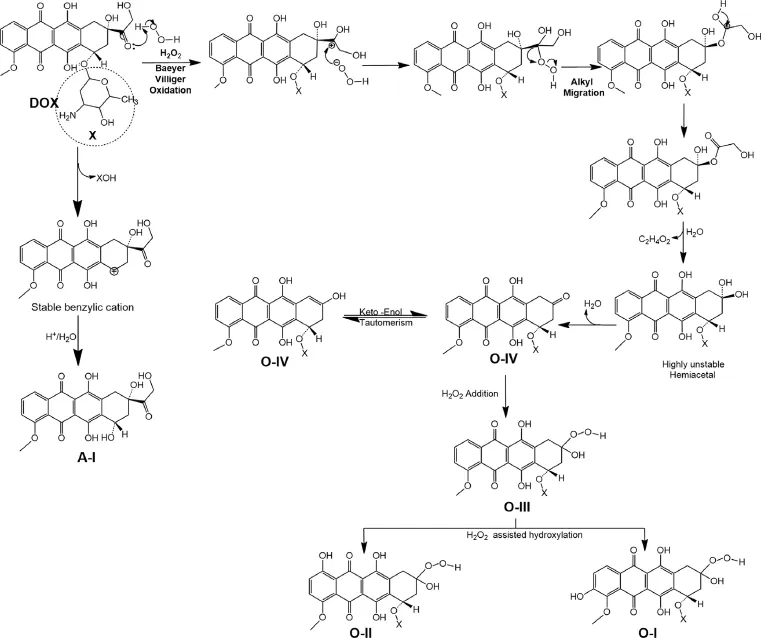
Fig.7.Mechanism of degradation of DOX.
The LC–MS–TOF spectra of O-II and O-I showed major and heaviest peaks atm/z534.1595 and 534.1601,hence they were assigned as parent ions(MIIand MI,respectively)(Fig.5D and E). An even mass of MIand MIIsuggested an odd number of nitrogen atoms in the products[23],which indicated the single nitrogen atom in the amino sugar component in DOX to be intact in both O-I and O-II.The very similar values of MIand MII(Tolerance of 0.7% as per Elemental Composition Calculator Software)indicated that both O-I and O-II have the same chemical formula.However,O-I and O-II eluted at different retention timesi.e.6.9 min and 7.8 min, respectively(Fig.2),which suggested these products have different structures.The TOF spectra of both products displayed the same fragment ions(Fig.5D and E).This comparative information on chromatographic and mass spectral data of O-I and O-II suggested that though they have similar chemical formula,they have different structures,which may be isomers.The TOF spectrum of O-II showed fragment ions ofm/z500.1532,387.0728,371.0736,353.0636 and 325.0694(Fig.5D).The heaviest fragment(m/z500.1532)was 34.0063 Da less than that of MII,and this mass difference was found to correspond to an H2O2molecule(34.0055 Da). The same fragment was also noted as the heaviest fragment in TOF spectrum of O-III,wherein it was proposed to form by the loss of a water molecule(Fig.6).Moreover,MIIIwas also found to fragment tom/z484.1603(MIV)by the loss of an H2O2molecule(Fig.6).These comparisons in mass spectral data indicated that hydroxyhyroperoxide moiety is present in MII,similarly as in MIII,which implied that O-II is a derivative of O-III itself.Further,the mass difference of 15.9958 Da between MIIIand MIIwas found to correspond to an oxygen atom(15.9949 Da).It suggested that O-II might form by the addition of an oxygen atom in MIIIunder the oxidative condition, which was possible through peroxide catalyzed oxidation of ring A leading to the formation of a hydroxylated(phenol)derivative.Further,this hydroxylation in ring A could occur at positionorthoorparawith respect to methoxy group(activating group).A logical co-relation among chromatographic elution data and comparative mass spectral data of O-I and O-II,and possibility of hydroxylation in ring A of DOX suggested that O-I and O-II might be positional isomers(orthoandpara)of the hydroxylated derivative of O-III.As O-II eluted after O-I(Fig.2),O-I might be more polar than O-II.This co-relation between polarity and elution order was supported by the observation that elution order of O-III,O-IV and DOX was in concordance with their lipophilicity,determined as ClogPby ChemBioDraw Ultra 12.0 software(Table 1).The possibleorthohydroxyl derivative of O-III was predicted to be more polar than theparaisomer(Table 1).Hence,O-I was proposed to be anorthoisomer(o-anisole derivative)whereas O-II to be theparaisomer(p-anisol derivative),which underwent mass fragmentation in agreement with their mass spectral data (Fig.6).Hence,O-I and O-II were suggested to be 3-hydroxy-9-desacetyldoxorubicin-9-hydroperoxide and 1-hydroxy-9-desacetyldoxorubicin-9-hydroperoxide,respectively.
3.3.4.Product A-I
It was detected as the heaviest ion peak atm/z415.1024(Fig.5F), which was assigned as its parent ion(MA)peak.An odd molecular mass of MAindicated an even number nitrogen or no nitrogen atom [23],which suggested that amino sugar moiety might be absent in A-I. MAwas also noted as a product ion in MS2spectrum of DOX,wherein it was proposed to form by the loss of amino sugar moiety(X-H)from the parent ion of DOX.Based on this,A-I was proposed to be deglucosaminyl doxorubicin formed by the loss of the glucosamine moiety due to cleavage of glycosidic linkage between the tetracycline ring and the glucosamine moiety.Mass fragmentation of the proposed A-I to product ions ofm/z397.0914,379.0824,361.0781 and 321.0745(Fig.4) was in agreement with its TOF spectrum.
3.4.Drug degradation mechanisms
Degradation of DOX to deglucosaminyl doxorubicin(A-I)under acid hydrolytic condition was possible through well known acid catalyzed cleavage of glycosidic linkage(Fig.7)similar as the reports on the degradation of other similar drug molecules such as idarubicin,doxorubicin,4′-deoxydoxorubicin,4′-O-methyl doxorubicin,4′-epidoxorubicin,doxorubininol,daunorubicin and carminomycin under acidic conditions[14,25].Degradation of DOX to O-IV(9-desacetyldoxorubicin)and then to O-III(9-desacetyldoxorubicin-9-hydroperoxide)was possible through Baeyer Villiger oxidative deacetylation following the addition of H2O2(Fig.7), similar to the oxidative degradation of idarubicin[25].The products O-I(3-hydroxy-9-desacetyldoxorubicin-9-hydroperoxide)and O-II(1-hydroxy-9-desacetyldoxorubicin-9-hydroperoxide)were proposed to form by peroxide assisted hydroxylation in ring A of doxorubicin.This proposition was supported by a report that disclosed and discussed the formation of alkoxy and aryloxy substituted phenols from the corresponding alkoxy and aryloxy benzene[26].However,a comparison of the results of the present study with degradation behavior of idarubicin reported earlier by our laboratory[25]revealed that such hydroxylated products did not form from idarubicin.This difference in degradation behaviors of doxorubicin and idarubicin was attributed to the methoxy group in ring A in doxorubicin,which is a moderate activating group and directs substitution atorthoandparaposition[27].
4.Conclusion
DOX was subjected to forced degradation under the conditions of hydrolysis(water,acid and alkali),peroxide oxidation,dry heat and photolysis as recommended in ICH guidelines Q1(R2).DOX degraded to several degradation products in alkaline medium even at room temperature that could not be resolved.Four degradation products (O-I–O-IV)were formed in oxidative conditions,and a single product (A-I)was formed in acid hydrolytic condition.They were resolved by a single isocratic LC–UV method and characterized by MS6and LC–MS–TOF studies as 3-hydroxy-9-desacetyldoxorubicin-9-hydroperoxide(O-I),1-hydroxy-9-desacetyldoxorubicin-9-hydroperoxide(OII),9-desacetyldoxorubicin-9-hydroperoxide(O-III),9-desacetyldoxorubicin(O-IV),and deglucosaminyl doxorubicin(A-I).While O-I–OIV were found to be new degradation impurities,A-I was found to be a pharmacopoeial impurity of DOX.The drug remained stable to hydrolytic degradation in neutral medium,thermal degradation and photolytic degradation.
Acknowledgments
The authors are thankful to Strides Arcolabs Pvt.Ltd.(Bangalore,India)for providing DOX as generous gift sample and to Prof. Saranjit Singh,Head,Department of Pharmaceutical Analysis,National Institute of Pharmaceutical Education and Research(SAS Nagar,India)for extending the facilities to carry out photostability and mass spectral studies.The authors are also thankful to All India Council for Technical Education(AICTE),New Delhi,India for providing f nancial assistance for carrying out the project(File No. 8-85/RIFD/RPS/Policy-3/2013-14).
Appendix A.Supporting information
Supplementary data associated with this article can be found in the online version at http∶//dx.doi.org/10.1016/j.jpha.2015.05.003.
[1]Guidance for Industry,ANDAs,Impurities in Drug Substances,Center for Drug Evaluation and Research Food and Drug Administration,MD,USA, 2009.
[2]International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use(ICH),Guidelines Q3C (R4)∶Impurities∶Guidelines for Residual Solvents,Geneva,Switzerland, 2009.
[3]International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use(ICH),Guidelines Q3D∶Impurities∶Guidelines for Elemental Impurities,Geneva,Switzerland,2013.
[4]International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use(ICH),Guidelines Q3A(R2)∶Impurities in New Drug Substances,Geneva,Switzerland,2006.
[5]International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use(ICH),Guidelines Q3B(R2)∶Impurities in New Drug Products,Geneva,Switzerland,2006.
[6]International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use(ICH),Guidelines Q1A(R2)∶Stability Testing of New Drug Substances and Products,Geneva,Switzerland,2003.
[7]L.A.Mitscher,A.Dutta,Antitumor natural products,in∶sixth ed.,in∶D.J.Abraham(Ed.),Berger’s Medicinal Chemistry and Drug Discovery,vol.5, John Wiley&Sons,New Jersey,2003,pp.110–128.
[8]G.Zogotto,B.Gatto,S.Moro,et al.,Anthrcyclines∶recent developments in their separation and quantitation,J.Chromatogr.B 764(2001)161–171.
[9]British Pharmacopoeia Commission,British Pharmacopoeia,vol.1,The Stationary Of f ce,London,2008.
[10]The United States Pharmacopeial Convention,United State Pharmacopoeia, vol.2,Port City Press,Baltimore,2007.
[11]Indian Pharmacopoeia Commission,Indian Pharmacopoeia,vol.1,National Institute of Science Communication and Information Resources(NISCAIR), New Delhi,2007.
[12]M.Martindale,Martindale∶The Complete Drug Reference,thirty third ed., Royal Pharmaceutical Society,London,2005.
[13]J.Cielecka-Piontek,A.Jelińska,M.Zając,et al.,A comparison of stability of doxorubicin and daunorubicin in solid state,J.Pharm.Biomed.Anal.50(2009) 576–579.
[14]J.H.Beijnen,G.Wiese,W.J.M.Underberg,Aspects of the chemical stability of doxorubicin and seven other anthracyclines in acidic solution,Pharm.Weekbl. 7(1985)109–116.
[15]M.J.Wood,W.J.Irwin,D.K.Scott,Stability of doxorubicin,daunorubicin and epirubicin in plastic minibags and plastic syringes,J.Clin.Pharm.Ther.15 (1990)279–289.
[16]M.J.Wood,W.J.Irwin,D.K.Scott,Photodegradation of doxorubicin,daunorubicin and epirubicin in f uorescent light and sunlight using HPLC,J.Clin. Pharm.Ther.15(1990)291–300.
[17]K.Nawara,P.Krysinski,G.J.Blanchard,Photoinduced reactivity of doxorubicin∶catalysis and degradation,J.Phys.Chem.A 116(2012)4330–4337.
[18]A.Ramu,M.M.Mehta,J.Liu,et al.,The ribo f avin mediated photooxidation of doxorubicin,Cancer Chemother.Pharmacol.46(2000) 449–458.
[19]A.Ramu,M.M.Mehta,T.Leaseburg,et al.,The enhancement of ribo f avin mediated photooxidation of doxorubicin by histidine and urocanic acid,Cancer Chemother.Pharmacol.47(2001)338–346.
[20]L.Bomgaars,S.Gunawardena,S.E.Kelley,et al.,The inactivation of doxorubicin by long ultraviolet light,Cancer Chemother.Pharmacol.40(1997) 506–512.
[21]International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use Stability Testing,Photostability Testing of New Drug Substances and Products,ICH Q1B,Geneva, Switzerland,1998.
[22]L.Sleno,V.C.Slate,D.A.Volmer,Dissociation reactions of protonated anthracycline antibiotics following electrospray ionization-tandem mass spectrometry,Int.J.Mass Spectrom.255–256(2006)130–138.
[23]S.Singh,T.Handa,M.Narayanam,et al.,A critical review on the use of modern sophisticated hyphenated tools in the characterization of impurities and degradation products,J.Pharm.Biomed.Anal.69(2012) 148–173.
[24]R.M.Silverstein,F.X.Webster,Spectrometric Identi f cation of Organic Compounds,sixth ed.,John Wiley&Sons,New York,1998.
[25]D.Kaushik,G.Bansal,Characterization of degradation products of idarubicin through LC–UV,MSnand LC-MS-TOF studies,J.Pharm.Biomed.Anal.85 (2013)123–131.
[26]M.Constantini,D.Lauchner,Catalytic Hydroxylation of Phenols/Phenol Ethers, U.S.Patent No.50970 78,1992.
[27]R.T.Morrison,R.N.Boyd,Organic Chemistry,sixth ed.,Prentice Hall of India, New Delhi,1992.
☆Peer review under responsibility of Xi'an Jiaotong University.*
E-mail addresses:gulshanbansal@rediffmail.com,
gulshanbansal@gmail.com(G.Bansal).
http∶//dx.doi.org/10.1016/j.jpha.2015.05.003
2095-1779/©2015 Xi'an Jiaotong University.Production and hosting by Elsevier B.V.All rights reserved.This is an open access article under the CC BY-NC-ND license (http∶//creativecommons.org/licenses/by-nc-nd/4.0/).
 Journal of Pharmaceutical Analysis2015年5期
Journal of Pharmaceutical Analysis2015年5期
- Journal of Pharmaceutical Analysis的其它文章
- Species authentication and geographical origin discrimination of herbal medicines by near infrared spectroscopy∶A review☆
- Multiple responses optimization in the development of a headspace gas chromatography method for the determination of residual solvents in pharmaceuticals☆
- Application of RP–HPLC method in dissolution testing and statistical evaluation by NASSAM for simultaneous estimation of tertiary combined dosages forms☆
- Determination of atractylon in rat plasma by a GC–MS method and its application to a pharmacokinetic study☆
- Rapid screening and distribution of bioactive compounds in different parts ofBerberis petiolarisusing direct analysis in real time mass spectrometry☆
- High-sensitivity simultaneous liquid chromatography–tandem mass spectrometry assay of ethinyl estradiol and levonorgestrel in human plasma☆