
Waldenstrom′s macroglobulinemia associated with Hodgkin′s lymphoma: a case report
2015-01-10 01:46YuanFuHuayuanZhuPengLiu
Yuan Fu, Huayuan Zhu, Peng Liu
Department of Hematology, the First Affiliated Hospital, Nanjing Medical University, Nanjing, Jiangsu 210029, China.
Waldenstrom′s macroglobulinemia associated with Hodgkin′s lymphoma: a case report
Yuan Fu△, Huayuan Zhu△, Peng Liu
Department of Hematology, the First Affiliated Hospital, Nanjing Medical University, Nanjing, Jiangsu 210029, China.
Waldenstrom′s macroglobulinemia/lymphoplasmacytic lymphoma (WM/LPL) is a low-grade B-cell non-Hodgkin′s lymphoma with an indolent clinical course. Higher-grade non-Hodgkin lymphoma (NHL) and therapyrelated myelodysplasia/acute leukemia (t-MDS/AML) have been reported in patients with WM/LPL in previous studies. However, only two cases with WM/LPL were reported to develop to Hodgkin lymphoma (HL). Here, we report the first case of WM/LPL who developed classical HL simultaneously 3 years after initial nucleoside analog-based chemotherapy.
Waldenstrom′s macroglobulinemia, Hodgkin′s lymphoma, Richter′s syndrome
Introduction
Waldenstrom′s macroglobulinemia/lymphoplasmacytic lymphoma (WM/LPL) is a lymphoproliferative disease characterized by an IgM gammopathy and accounts for approximately 2% of all hematological malignant neoplasms. It has an indolent course and the main causes of death are bone marrow hematopoietic failure, infection, embolism and heart failure. WM/LPL patients have a median age of 65 years and a male predominate with a ratio of male:female of 2:1[1]. Transformation to higher-grade non-Hodgkin lymphoma (NHL) and therapy-related myelodysplasia/acute leukemia (t-MDS/AML) have been described in case reports and small series[2-4]. However, only two reports of patients with WM transformed to Hodgkin disease[2]. In this study, we reported the first case in China with a patient who was initially diagnosed as WM/LPL and then presented with WM/LPL and classical Hodgkin′s lymphoma (HDHL) simultaneously 3 years later.
Case report
A 74-year-old man was admitted to our department in June 2011 with the diagnosis of WM. He had fatigue, dizziness, night sweat and enlargement of left cervical lymph nodes for approximately 2 months. Laboratory examinations in 2008 revealed hemoglobin at 94 g/L (reference range, 110 g/L-160 g/L) and IgM at 46.9 g/L (reference range, 0.4 g/L-2.3 g/L) and serum immunofixation electrophoresis showed an IgM-kappa monoclonal peak. Bone marrow aspiration and biopsy confirmed the diagnosis of WM with the appearance of lymphoid plasma cells. The patient was diagnosed as WM and treated with 3 courses of fludarabine andcyclophosphamide (FC) from October 2008 to December 2008. He achieved complete remission with the disappearance of malignant lymphoid cells in the bone marrow and normal IgM immunoglobulin level. However, he has not been followed up closely in the next 2 years. He developed giddiness and fatigue in April 2011, and subsequently headache, pain on the left neck, blurred vision, night sweat and fever in the following weeks, and was admitted to our department. Physical examination revealed enlarged lymph nodes in the supraclavicular, anterior cervical and submandibular areas, but no hepatosplenomegaly. Laboratory examinations were shown as follows: Complete blood count revealed white blood cells (WBC) at 5.1×109/L (reference range, 3.9-10.0×109/L), hemoglobin at 90 g/L, and platelets at 316×109/L (reference range 100-300×109/L), and IgM at 31.8 g/L. Serum immunofixation electrophoresis still showed an IgM-kappa monoclonal peak. Other tests included: elevated serum β-2 microglobulin (4.07 mg/L; reference range, 0.40 mg/L-0.49 mg/L), increased erythrocyte sedimentation rate (ESR) (125 mm/H; reference range, 0.00-40.00 mm/H),and high serum content of κ-light chain (6.45 g/L; reference range: 1.7-3.7 g/L). Karyotype analysis showed 46, XY, and the test for Ig H rearrangement was positive. Flow cytometry did not find CD45-CD138+CD56+CD19- cells in bone marrow. Bone marrow aspiration showed infiltration of lymphoid plasma cells (Fig. 1). However, lymph node biopsy revealed that the lacunar cells were positive for CD30, CD15, CD20 and PAX-5 and negative for LCA, consistent with the diagnosis of classical Hodgkin′s lymphoma (mixed cell type) (Fig. 2). Moreover, PET/CT confirmed the enlargement of several lymph nodes in the cervical and submandibular region and a tumoral mass in posterior wall of nasopharynx of the size of 1.6×1.3 cm, and the highest standardized uptake value (SUV) was 11.2 in all involved sites. Emission computed tomography (ECT) did not reveal any bone lesion. The patient was then diagnosed as classical Hodgkin′s lymphoma, mixed cell type with WM. He received the ABVD regime (vinblastine, dacarbazine, bleomycin and doxorubicin) since June, 2011. Fever, blurred vision and night sweat were relieved, and cervical lymph nodes were reduced 30% in volume after one cycle of treatment. The dose of doxorubicin was reduced by 75% due to atrial premature beats on a 24-hour Holter electrocardiogram. Unfortunately, his symptom progressed 1 month after the second cycle of the ABVD regimen. The patient wastreated with the R-Gemox (decitabine and oxaliplatin) regimen as salvage chemotherapy. However, he did not respond to chemotherapy and was complicated with sepsis, and finally succumbed to respiratory failure on November 11, 2011.
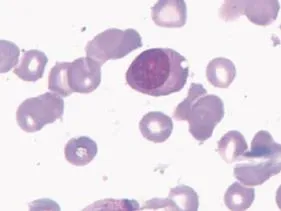
Fig. 1 Lymphoid plasma cells in bone marrow aspiration specimen (Wright-Giemsa, ×100).
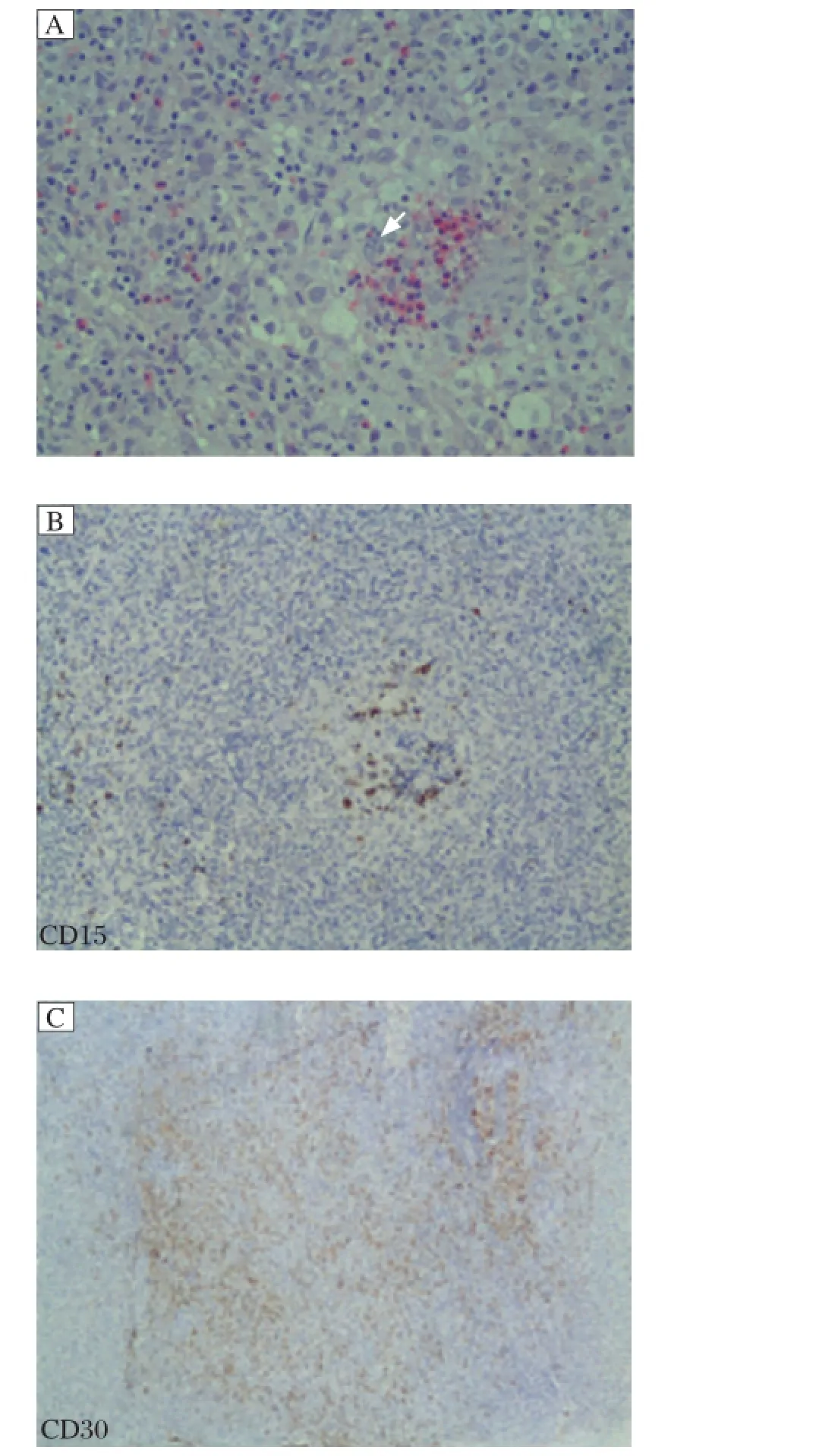
Fig. 2 Left supraclavicular lymph node biopsy specimen (H&E, ×400). A: Many lacunar cells and scant Reed-Sternberg (RS) cells are found. Arrow indicates RS cell. B (CD15 positive) and C (CD30 positive) show the lacunar cells of left supraclavicular lymph node biopsy specimen.
Discussion
WM is a distinct B-cell disorder, which is characterized by the accumulation of clonally lymphoplasmacytic cells mainly infiltrating the bone marrow and secreting a monoclonal IgM protein. WM is considered lymphoplasmacytic lymphoma (LPL) by the World Health Organization (WHO) classification system. The majority of LPL/WM patients have an indolent course. Previous studies have reported that patients may develop higher-grade NHL, such as diffuse large B cell lymphoma (DLBCL), therapyrelated myelodysplasia/acute leukemia (t-MDS/AML) and immunoblastic sarcoma, especially those patients who have been previously treated with nucleoside analogs[3-4]. However, only two cases of LPL/WM who developed to HL have been reported[2]. Here, we report a patient initially diagnosed as LPL/WM and presented with LPL/WM and HL simultaneously 3 years later. Our patient was diagnosed as WM in 2008 confirmed by the presence of monoclonal IgM detected on serum protein electrophoresis and the appearance of lymphoid plasma cells in bone marrow aspirate and biopsy. He achieved complete remission after 3 courses of the FC regimen. Unfortunately, his disease progressed 3 years later. Lymph node biopsy revealed large cells with the immunochemistry of HL (mixed cell type). Interestingly, lymphoid plasma cells but not malignant large cells were found in his bone marrow and monoclonal IgM can also be detected on serum protein electrophoresis. Therefore, we defined that the patient had LPL/WM and HL simultaneously, maybe in the process of transformation from LPL/ WM to HL. He died of sepsis and disease progression 5 months after transformation.
Patients with chronic lymphocytic leukemia/small lymphocytic lymphoma who transform to a highgrade lymphoma, such as HL, hairy cell leukemia and NHL, especially DLBCL are described as Richter′s syndrome. Similarly, patients with LPL/WM may also develop high-grade lymphoma. Although the exact pathogenesis of Richter′s syndrome is still unclear, many mechanisms have been raised, including transformation triggered by viral infections, such as Epstein-Barr virus (EBV)[5], the expression of genes that inhibit cancer development and chromosomal abnormalities. These abnormalities mainly include trisomy 12, structural abnormalities at 11q and mutations in tumor suppressor genes, especially in p53 and p16INK4A[6]. EBV can be detected in Reed-Sternberg cells of about 35% of HD patients, but whether EBV can cause HD was not confirmed[7-8]. In our case, the patient was negative for EBV transcription and had a normal karyotype, which may suggest that EBV infections and the expression of genes implicated in cancer development and chromosomal abnormalities were probably not involved in the pathogenesis of LPL/WM transforming to HL. Leleu et al. reported an increased incidence of transformation to high-grade NHL and the development of t-MDS/AML among patients with WM treated with nucleoside analogs[3]. Ling et al. also reported two cases of WM with unusually aggressive transformation with treatment of cladribine[9]. However, Lin et al. reported an incidence of 13% among 92 patients who developed DLBCL, and these patients were associated with poor prognosis and EBV was not involved in the pathogenesis of transformation[4]. Although risk factors for developing the transformation in patients with LPL/WM are poorly understood at present, the transformation of our patient may be due to the use of nucleoside analogs-based chemotherapy (the FC regimen) at the initial treatment.
In summary, we report a rare case who presented with HL and LPL/WM simultaneously 3 years after the initial diagnosis of LPL/WM. Evolution to HL may be attributed to exposure to nucleoside analogs.
[1] Dimopoulos MA, Panayiotidis P, Moulopoulos LA, et al. Waldenström′s macroglobulinemia: clinical features, complications, and management[J]. J Clin Oncol, 2000, 18(1):214-226.
[2] Rosales CM, Lin P, Mansoor A, et al. Lymphoplasmacytic lymphoma/Waldenstrom macroglobulinemia associated with Hodgkin disease. A report of two cases[J]. Am J Clin Pathol, 2001, 116(1):34-40.
[3] Leleu X, Soumerai J, Roccaro A, et al. Increased incidence of transformation and myelodysplasia/acute leukemia in patients with Waldenstrom macroglobulinemia treated with nucleoside analogs[J]. J Clin Oncol, 2009, 27(2):250-255.
[4] Lin P, Mansoor A, Bueso-Ramos C, et al. Diffuse large B-cell lymphoma occurring in patients with lymphoplasmacytic lymphoma/Waldenstrom macroglobulinemia. Clinicopathologic features of 12 cases[J]. Am J Clin Pathol, 2003, 120(2):246-253.
[5] Ansell SM, Li CY, Lloyd RV, Phyliky RL. Epstein-Barr virus infection in Richter′s transformation[J]. Am J Hematol, 1999, 60(2):99-104.
[6] Rossi D, Cerri M, Capello D, et al. Biological and clini-cal risk factors of chronic lymphocytic leukaemia transformation to Richter syndrome[J]. Br J Haematol, 2008, 142(2):202-215.
[7] Omoti CE, Omoti AE. Richter syndrome: a review of clinical, ocular, neurological and other manifestations[J]. Br J Haematol, 2008, 142(5):709-716.
[8] Nourse J, Jones K, Gandhi MK. The role of Epstein-Barr virus in Richter syndrome[J]. Br J Haematol, 2009, 144(4):613; author reply 614-615.
[9] Ling S, Joshua DE, Gibson J, et al. Transformation and progression of Waldenstrom′s macroglobulinemia following cladribine therapy in two cases: natural evolution or iatrogenic causation?[J]. Am J Hematol, 2006, 81(2):110-114.
△These authors contributed equally to this work.
Tel/fax: +86-25-83781120/+86-25-83781120, E-mail: liupeng8888@ yahoo.com.cn.
Received 24 December 2012, Accepted 12 March 2013, Epub 06 June 2013 The authors reported no conflicts of interest.
R552.1, Document code: B
© 2015 by the Journal of Biomedical Research. All rights reserved.
10.7555/JBR.27.20120144
 THE JOURNAL OF BIOMEDICAL RESEARCH2015年6期
THE JOURNAL OF BIOMEDICAL RESEARCH2015年6期
- THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- Platelets in hemostasis and thrombosis:Novelmechanisms of fibrinogen-independent platelet aggregation and fibronectinmediated protein wave of hemostasis
- Retinolbinding protein 4 correlates with and is an early predictor of carotid atherosclerosis in type 2 diabetes mellitus patients
- CC-chemokine receptor 7 and its ligand CCL19 promote mitralvalve interstitialcellmigration and repair
- Effectofburden and origin sites ofpremature ventricular contractions on left ventricular function by 7-day Holter monitor
- Prevention ofatrialfibrillation with renin-angiotensin system inhibitors on essentialhypertensive patients:a meta-analysis of randomized controlled trials
- Epirubicin-gold nanoparticles suppress hepatocellular carcinoma xenograft growth in nude mice