
CC-chemokine receptor 7 and its ligand CCL19 promote mitralvalve interstitialcellmigration and repair
2015-01-10 01:46XiaozhiWangLiangWangLipingMiaoRongZhaoYanhuWuXiangqingKong
Xiaozhi Wang,Liang Wang,Liping Miao,Rong Zhao,Yanhu Wu,Xiangqing Kong
1Department of Cardiology;2Department of Cardiothoracic Surgery,The First Affiliated Hospital of Nanjing Medical University,Nanjing,Jiangsu 210029,China.
CC-chemokine receptor 7 and its ligand CCL19 promote mitralvalve interstitialcellmigration and repair
Xiaozhi Wang1,△✉,Liang Wang1,△,Liping Miao1,Rong Zhao1,Yanhu Wu2,Xiangqing Kong1
1Department of Cardiology;2Department of Cardiothoracic Surgery,The First Affiliated Hospital of Nanjing Medical University,Nanjing,Jiangsu 210029,China.
The effectof CC-chemokine receptor 7(CCR7)and CC-chemokine ligand 19(CCL19)on rheumatic mitralstenosis is unknown.This study aimed to explore the roles of CCR7 and CCL19 in rheumatic mitralstenosis by measuring the expression of CCR7 and CCL19 in human mitral valves from rheumatic mitral stenosis patients. Additionally,we examined their effects on human mitralvalve interstitialcells(hMVICs)proliferation,apoptosis and wound repair.CCR7 and CCL19 expression was measured in the mitralvalves from rheumatic mitralstenosis patients(n=10)and compared to normalmitralvalves(n=5).CCR7 was measured in cultured hMVICs from rheumatic mitralstenosis patients and normaldonors by RT-PCR and immunofluorescence.The cells were also treated with exogenous CCL19,and the effects on wound healing,proliferation and apoptosis were assayed.In the rheumatic mitralvalves,valve interstitialcells expressed CCR7,while mononuclearcellsand the endothelium expressed CCL19.Healthy mitralvalves did notstain positive for CCR7 or CCL19.CCR7 was also detected in cultured rheumatic hMVICs or in normalhMVICs treated with CCL19.In a wound healing experiment,wound closure rates of both rheumatic and normalhMVICs were significantly accelerated by CCL19.These effects were abrogated by a CCR7 neutralizing antibody.The CCR7/CCL19 axis did notinfluence the proliferation or apoptosis of hMVICs, indicating thatwound healing was due to increased migration rates rather than increased proliferation.In conclusion,CCR7 and CCL19 were expressed in rheumatic mitralvalves.The CCR7/CCL19 axis may regulate remodeling of rheumatic valve injury through promoting migratory ability of hMVICs.
CC-chemokine receptor 7,CC-chemokine ligand 19,rheumatic mitral stenosis,migration,wound repair
Introduction
Although the prevalence of rheumatic fever has greatly decreased in developed countries,the late sequelae ofrheumatic fever,rheumatic mitralstenosis, stillresults in high morbidity and mortality in developing countries[1].While long-term penicillin treatment can effectively decrease the recurrence of rheumatic fever,it cannot prevent mitral valve pathology that results afterrheumatic fever[2].In response to rheumatic fever,an autoimmune response in the mitral valve damages the endothelial layer,which triggers a subsequent inflammatory response thatinvolves neovascu-larization[3],stimulating lymphocyte infiltration into the valve.Even in aged calcified rheumatic valves,lymphocytes and pro-inflammatory cytokines,as well as increased endothelialization,are still present,indicating a persistent state of inflammation in the diseased valves[4-5].
CC-chemokine ligand 19(CCL19)and its corresponding receptor,CC-chemokine receptor7(CCR7),regulate homing of T lymphocytes and dendritic cells[6].In addition to a role in inflammatory celltrafficking,CCR7 has been implicated in metastatic breast cancer[7]and tissue repair[8].CCR7 and CCL19 are expressed by mast cells,airway smooth muscle cells,myofibroblasts,and fibroblasts.Furthermore,CCR7 has been shown to mediate cellmigration in asthma[9]and is expressed by several mesenchymalcelllines.Valve interstitialcells are the predominant mesenchymalcell type found in the 3 layers of the heartvalve.In addition,valve interstitialcells are both fibroblasts and myofibroblasts,and are very important in control of heart valve homeostasis and diseased valve repair or remodeling[10-11].Since significant lymphocyte infiltration occursduring allthe stagesofrheumatic mitral stenosis[12-13],we hypothesized that CCR7 and CCL19 may regulate the pathologicalbehavior of human mitral valve interstitialcells(hMVICs),which are involved in chronic remodeling ofstenotic mitralvalves.In thisstudy, we investigated the expression of CCR7 and CCL19 in rheumatic mitralstenosis mitralvalves and whether the CRR7/CCL19 axis mediated hMVIC function.
Materials and methods
Ethics statement
This study was approved by the Ethics Committees ofthe authors'affiliated institution(Protocol No.2011-SRFA-057)and conformed to the principlesoutlined in the Declaration of Helsinki.Written consent was obtained from allparticipants ortheir parents involved in this study.
Sample collection,cell isolation and culture
Rheumatic stenotic mitralvalveswere obtained from 10 patients who underwentsurgicalvalve replacement for rheumatic mitral stenosis(3 males and 7 females with a mean age of48±7 years).Normalmitralvalves were obtained from 5 healthy donor hearts following the donors’deaths in traffic accidents(2 males and 3 females with a mean age of 37±6 years).
Normalvalves and non-calcific rheumatic stenotic valveswere used forcellisolation.hMVICswere isolated by collagenase digestion as previously described[14],and then were cultured in Dulbecco's modified eagle medium (DMEM;Hyclone,Logan,UT,USA)supplemented with 10%fetalbovine serum(FBS;Hyclone),100 U/mL penicillin,100 mg/mL streptomycin,and 4 mmol/L L-glutamine.Primary hMVICs were phenotypically examined by immunostaining with primary antibodies against vimentin(Abcam,ab45939)andα-smooth muscle actin (Cy3 conjugated,Sigma,St Louis,MO,USA,C6198). Following incubation with theprimary antibodies,sections were incubated with a secondary Dylight TM488-conjugated donkey anti-rabbit Ig G antibody(Jackson ImmunoResearch Laboratory,West Grove,PA,USA). Cellsfrom passages 3 to 5 were used forallexperiments.
Immunohistochemistry and immunofluorescence
Immunohistochemistry was performed using the ImmunoCruzTMmouse ABC Staining System(Santa Cruz Biotechnology,Santa Cruz,CA,USA,sc-2017). The same leaflets of the mitralvalve from the rheumatic mitralstenosisand normalgroupswere fixed in 4%paraformaldehyde for 17 hours and mounted in paraffin. Sections(4μm)were pre-incubated with 10%hydrogen peroxide afterre-hydration,and then treated with 10%normal goat serum after antigen retrieval(0.1%trypsin at 37°C for30 minutes).The sections were incubated with antibodies against CCR7(monoclonalmouse anti-CCR7 antibody;Santa Cruz Biotechnology,sc-73846)or CCL19(monoclonalmouse anti-human CCL19 antibody; R&D Systems,Abingdon,UK,MAB361)for90 minutes at37°C.Secondary antibodies were HRP conjugated donkey anti-mouse IgG or donkey anti-goat IgG(Santa Cruz Biotechnology).The slides were rinsed in PBS(pH 7.4) 3 times after each incubation step.Finally,the slides were counterstained with hematoxylin,mounted,and observed under the lightmicroscope.
For immunofluorescence,hMVICs were grown to confluence on glass coverslips,fixed in 4%formaldehyde for 10 minutes,lysed with 0.3%Triton X-100(Promega)for 10 minutes atroom temperature,and incubated with a mouse anti-CCR7 antibody overnightat4°C.The next day,hMVICs were incubated with a Dylight TM488-conjugated donkey anti-mouse IgG antibody(Jackson),and then counterstained with DAPI(Sigma).Staining was detected by fluorescentmicroscopy atsame exposure time and CCR7 positive cellnumberand totalcellnumberwere calculated using Image Pro Plus 6.0 software.
Quantitative PCR
Total RNA was extracted using TRIzol(Invitrogen, Carlsbad,CA,USA).RNA quality was assessed by formaldehyde agarose gel electrophoresis and was quantified spectrophotometrically.RNA was reverse transcribed using the Transcriptor Strand cDNASynthesis kit(Roche,Version 6.0).The following primers for CCR7 q-PCR were used:forward 5′-TGAGGTCACGGACGATTACAT-3′,reverse 5′-TGGAGGACAGTGAAGAAAACG-3′and the amplicon length was 143 bp.Real-time PCR was performed using 2x SYBR GreenⓇPCR Master Mix on an AB 7900 Real-time PCR system(Applied Biosystems, Foster City,CA,USA).Each sample was analyzed in triplicate,and target genes were normalized to the reference housekeeping gene glyceraldehyde-3-phosphate dehydrogenase(GAPDH).Fold differences were then calculated foreach treatmentgroup using normalized CT values for the control.PCR products were additionally assessed by agarose gel electrophoresis, and then analyzed spectrophotometrically.
Wound healing assays
hMVICs were grown in 6-well culture dishes.After reaching confluency,a flat cell scraper(width 1 mm) was used to create a linear wound across the confluent monolayer.The wounded monolayer was then washed 3 times with standard medium to remove cell debris. Immediately after wounding,cells were treated with 100 or 200 ng/mL CCL19(R&D,361-MI-025),with or without15μg/mL anti-CCR7 neutralizing antibody (R&D,MAB197).Cells were subsequently fixed with 4%paraformaldehyde 0,24 or 48 hours after wounding and CCL19 treatment.The width between the base of each wound edge was calculated as previously described[15]for assessment of migration rate.In all conditions,experiments with 3 sets of cultured dishes foreach treatmentwere conducted in atleasttriplicate.
Proliferation assay
Cellproliferation wasassessed using the CellTiter96ⓇAQueous One Solution Cell Proliferation Assay kit (Promega)according to the manufacturer's instructions. Briefly,cellswere seeded onto 3 microtiterplatesata density of3,000 cells per100μL perwell.After24 hoursof serum starvation,cells were incubated for24,48 or72 hours with recombinantCCL19(R&D,361-MI-025)at various concentrations.DMEMalone served as the negative control.MTS(3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) reagent(20μL)wasadded to each welland incubated at 37°Cfor3 hours.Absorbancewasrecorded at490nmwith a microplatereader(ELx800,Bioteke,Beijing,China).All experimentswere repeated 3 times.
Apoptosis assay
Cells were seeded onto 6-cm culture dishes.After24 hours of serum starvation,cells were incubated for 24 and 48 hours with CCL19(R&D,361-MI-025)at100 ng/mL and 200 ng/mL.DMEMalone served ascontrols. Then,cells were respectively harvested using trypsin/ EDTA,washed with PBS,resuspended in 1 mL binding buffer,and stained with 10μL annexin V-FITC and 10 μL propidium iodide(PI)atroom temperaturefor15 minutes(Biovision,CA,USA).Fluorescence of FITC and propidium iodide was analyzed using flow cytometry as previously described[16].
Statistical analysis
Statisticalanalysis was performed using SPSS 17.0. Data were expressed as mean±SD.For continuous variables,differences ofmean valuesbetween the RMS and the normalgroups were tested using Student’s t-test, and for categoricalvariables Chi-square testwas used to compare proportions.One-way ANOVA was performed to compare non-treated and treated experiments,and P<0.05 was considered statistically significant.
Results
Baseline characteristics of study patients
Diagnosis of 10 enrolled rheumatic mitral stenosis patients met the updated Jones criteria[17].The clinical characteristics of all patients are summarized in Table 1.The rate of atrial fibrillation was 80%.The mean valve orifice area and mean leftatrialdiameterwere 0.91 cm2±0.05 cm2and 48.3 mm±1.0 mm,respectively.
CCR7 and CCL19 expression in the rheumatic mitral valves
Allvalvesfrom patients with rheumatic mitralstenosis demonstrated marked hypercellularity.CCR7 and CCL19 were notobserved in explanted normalmitral valves(Fig.1A and C).Valve interstitialcells within the rheumatic valves expressed CCR7(Fig.1B). CCL19 was also highly expressed in the rheumaticvalves,butwas localized to the endothelium ofvessels, infiltrated mononuclear cells,and the extracellular matrix(Fig.1D-E).

Table 1 Baseline characteristics of the rheumatic mitral stenosis subjects
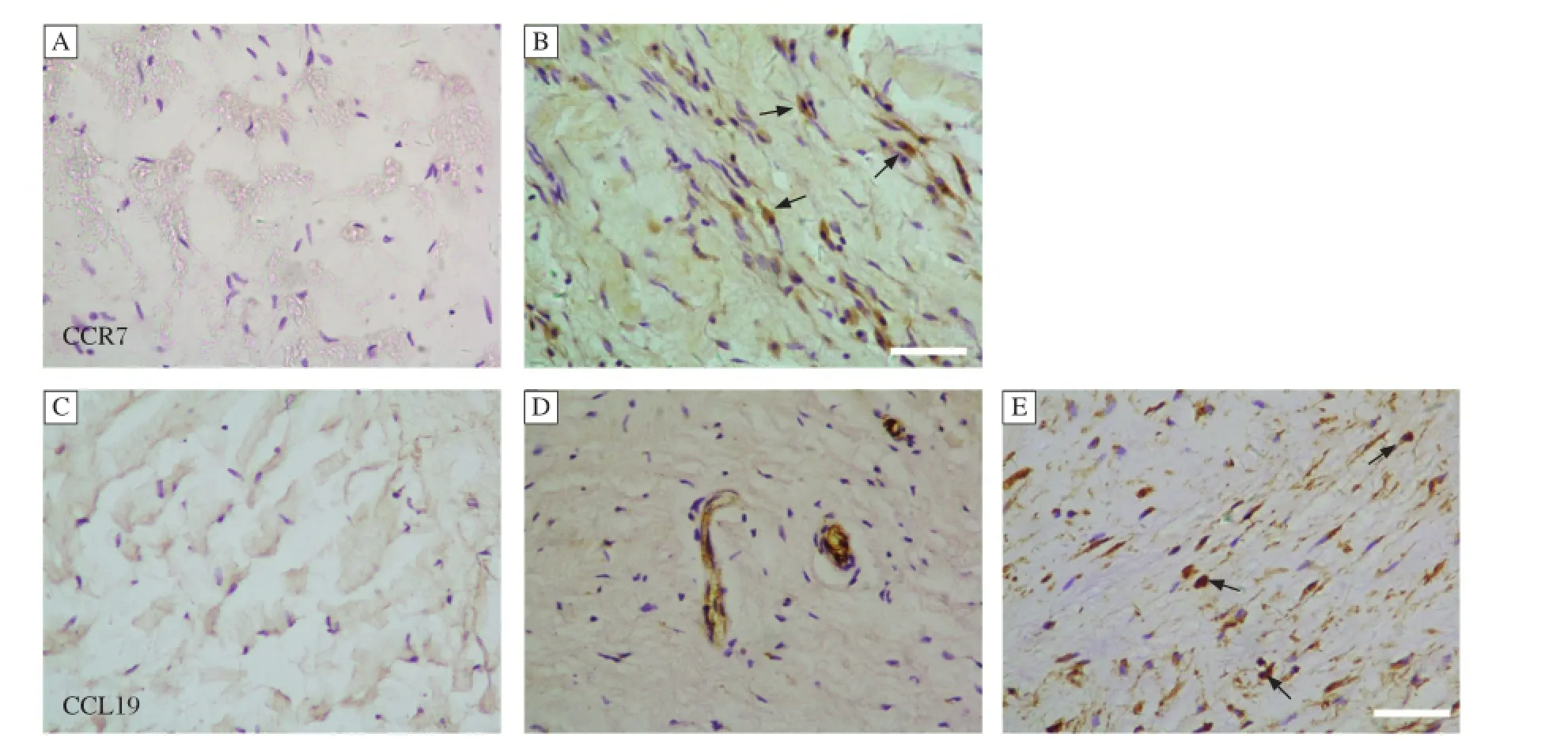
Fig.1 Representative photomicrographs of CCR7 and CCL19 expression in mitral valves.(A)CCR7 was not found in normal mitral valves.(B)CCR7 was expressed by hMVICs in mitralvalves of RMS(arrow).(C)CCL19 was notfound in normalmitralvalves.(D)CCL19 was also observed in the endothelium of vessels in mitralvalves of RMS.(E)CCL19 was expressed by mononuclear cells in mitralvalves of RMS(arrow).The scale bar is 50μM.
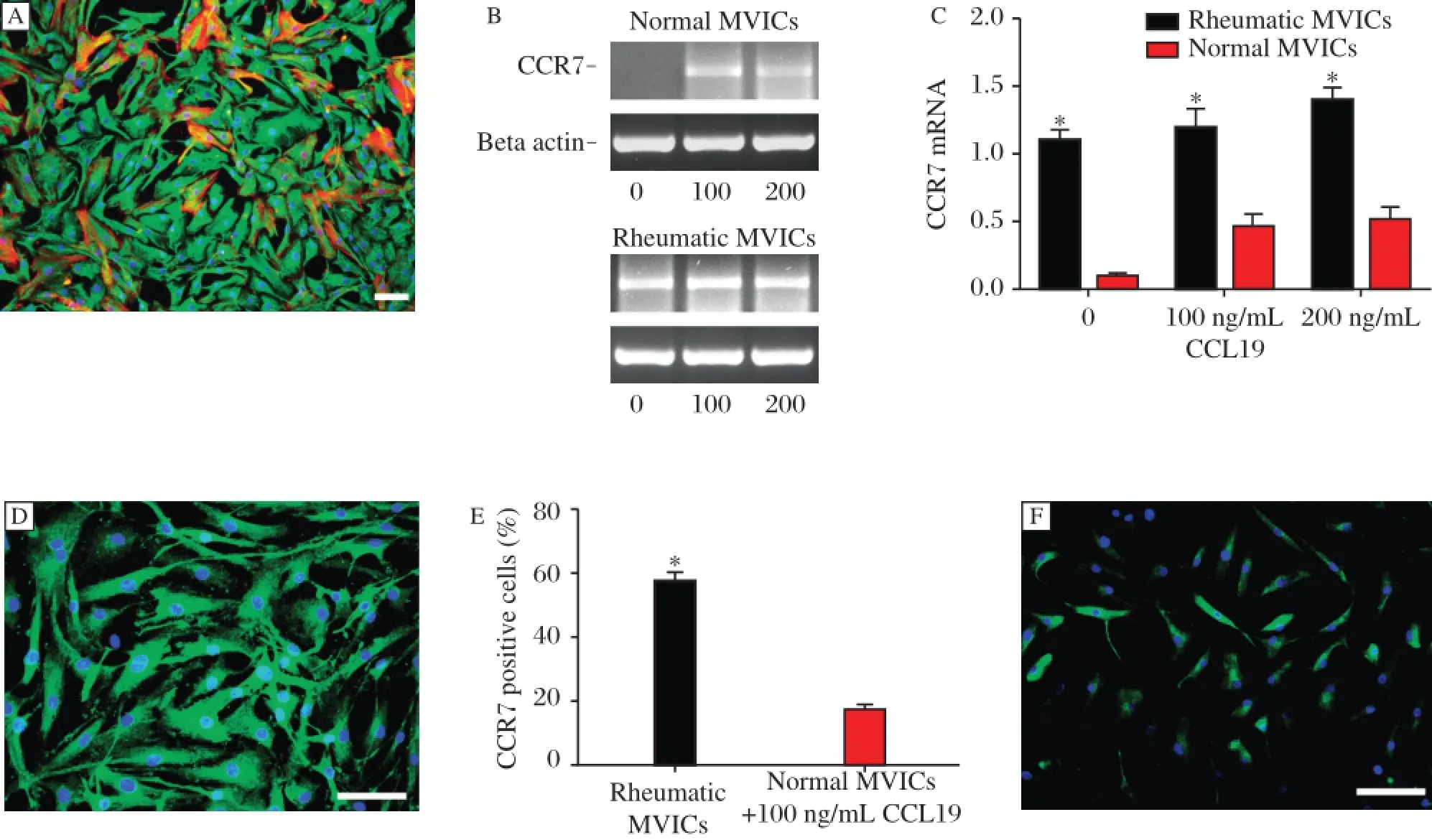
Fig.2 CCR7 expression in cultured hMVICs.(A)Representative photomicrograph of rheumatic hMVICs.Immnofluorescent staining showed that hMVICs consisted of vimentin positive(green)fibroblastic cells and vimentin and a-SMA double positive(red)myofibroblasts.(B)CCL19 stimulation was required before CCR7 was observed in normal hMVICs,while rheumatic hMVICs showed positive CCR7 staining withoutadditional CCL19 stimulation.(C)The CCR7 mRNA levelof rheumatic hMVICs was higherthan thatof normalhMVICs,butthe concentration of CCL19 did not alter the levelof CCR7 expression.*P<0.05.(D-F)CCR7 expression was confirmed by immunofluorescence in rheumatic hMVICs and 100 ng/ml CCL19 treated normal hMVICs.The more CCR7 positive cells were observed in rheumatic group even without CCL19 stimulation.*P<0.05.The imaging photos are captured at the same exposure time.The scale bar is 100μM.
CCR7 is expressed by cultured hMVICs in vitro
hMVICs isolated from the rheumatic mitralvalves were similar to those from normal valves.Both groups ofcells were positive forvimentin andα-SMA,consistentwith the appearance of spindle and polygon fibroblastormyofibroblastcells(Fig.2A).CCL19 was not expressed in cultured h MVICs from either the rheumatic or normal valves.
Quantitative PCR assays showed that CCR7 was expressed in rheumatic h MVICs,but not in normal hMVICs at baseline.Interestingly,normal hMIVCs expressed CCR7 after treatment with CCL19 (Fig.2B).CCR7 mRNA levelof rheumatic hMVICs was higher than thatof normal hMVICs(P<0.05, Fig.2C).Immunostaining also confirmed the PCR results,as the fluorescentintensity of CCR7 in hMIVCs from the rheumatic valves was more intense than from normalvalves(P<0.05,Fig.2D-F).
CCL19 promotes wound repair through CCR7 by stimulating migration
Wound healing response of hMVICs was significantly accelerated by treatment with recombinant CCL19.Both rheumatic and normal cells treated with CCL19 for48 hours showed a significantly greaterrate of wound closure than untreated cells(Fig.3A-I). hMVICs treated with a neutralizing antibody against CCR7,regardless of the addition of CCL19,did not show a significant difference in the extent of woundclosure compared with non-treated cells.The addition of CCL19 to hMVICs treated with a CCR7 neutralizing antibody resulted in a statistically reduced extent of wound closure,as compared to cells treated with CCL19 alone(Fig.4A-H).Interestingly,the addition of 200 ng/mL CCL19 accelerated wound closure at24 hours compared to cells treated with 100 ng/mL CCL19 in normalcells(Fig.4I).
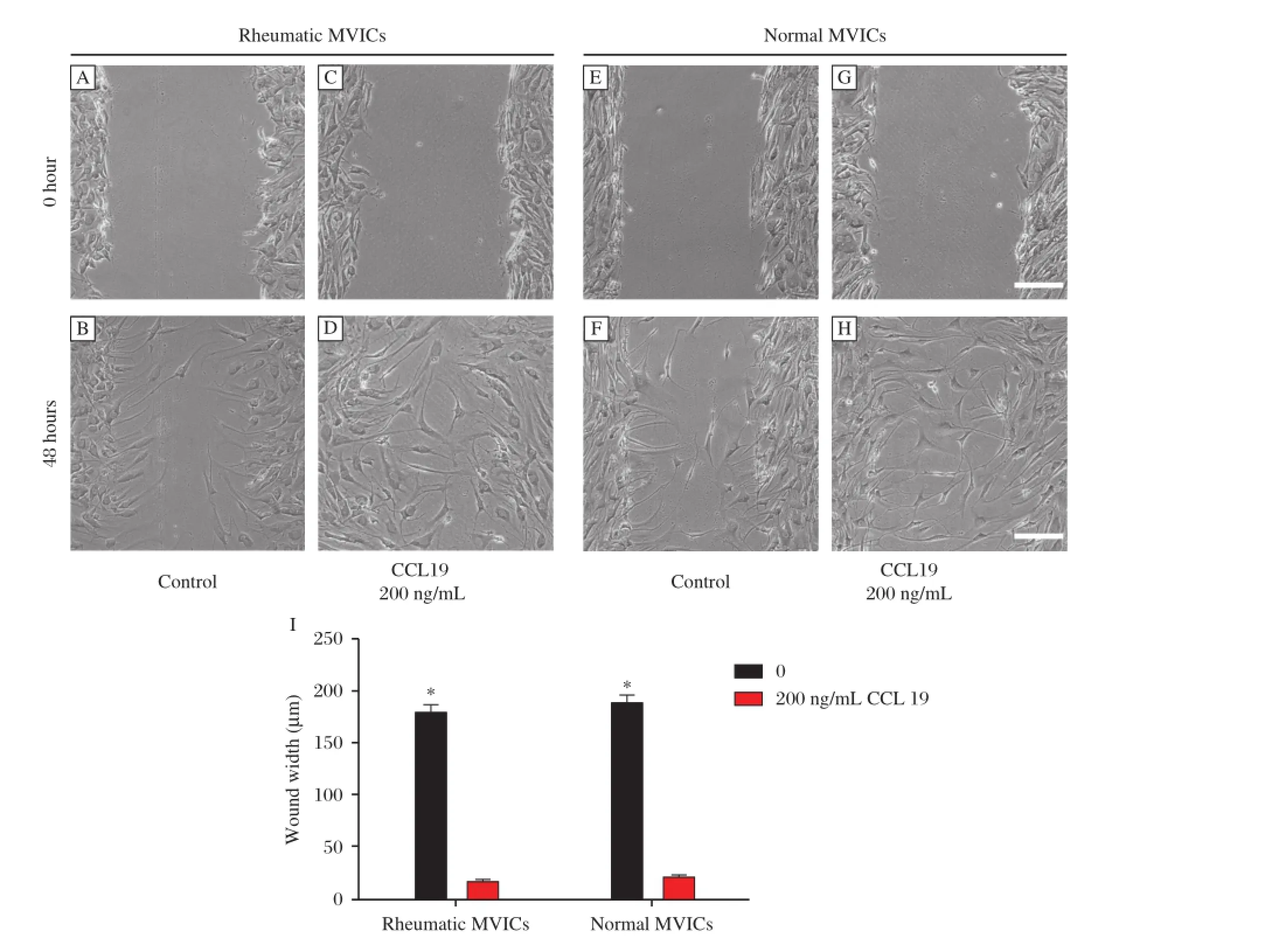
Fig.3CCL19 accelerated wound closure of the wounded hMVICs monolayer.(A-H)Representative photographs 48 h after wounding the hMVICs monolayer.(A-D,I)In rheumatic hMVICs,at48 h after wounding,the wound remained unclosed in hMVICs withouttreatment,butthe wound was sparsely closed and had no obvious gaps after 200 ng/ml CCL19 treatment.(E-H,I)In normalhMVICs,similar results with rheumatic hMVICs were observed.There were no significant differences between rheumatic and normalgroups.*P<0.05.The scale bar is 200μM.
Sincewound closureinvolvesboth cellproliferation and cellmigration,we measured cellproliferation to determine the impactofCCL19 on celldivision.The numberofcells, from both the rheumatic and normalvalves,cultured for 24,48 or72 hours in the presence of CCL19(1 ng/mL to 1,000 ng/mL)wasnotsignificantly differentcompared to cellscultured in mediaalone.Thisresultwasconfirmed with a MTS proliferation assay(Fig.5A-B).To exclude the impactof CCL19 associated apoptosis on cellgrowth and migration,we additionally measured cellapoptosis in the presence of CCL19(100 ng/mL and 200 ng/mL).The ratio of apoptotic cells in the CCL19-treated groups slightly decreased at 48 hours,butshowed no differences compared to the untreated grou ps(Fig.5C-D). Collectively,these results indicate that CCL19 exerted effects on migration,ratherthan proliferation or apoptosis, in wound healing.
Discussion
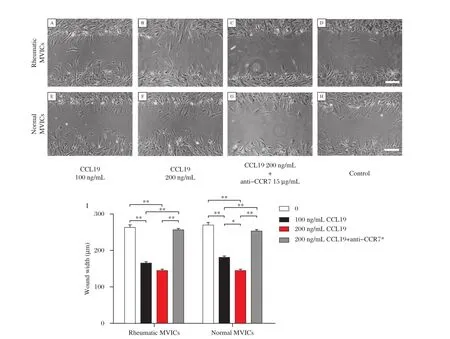
Fig.4 CCR7 dependent CCL19 promoted hMVICs migration.(A-H)Representative photomicrographs of hMVICs 24 h after wounding of the hMVICs monolayer.(A-D,I)In rheumatic groups,the migration rate of CCL19-treated hMVICs significantly increased compared with hMVICs cultured in media alone.There were no statistical differences between the 100 ng/ml and 200 ng/ml CCL19 groups.(E-I)In normal groups,the migration rate of CCL19-treated hMVICs significantly increased when compared with hMVICs cultured in media alone;however,the migration rate of cells treated with 200 ng/ml CCL19 was longer than cells treated with 100 ng/ml CCL19,indicating a dose response.The effect of CCL19 on migration was blocked with an anti-CCR7 neutralizing antibody.*15ug/ml anti-CCR7,*P<0.05,**P<0.01.The scale bar is 200μM.
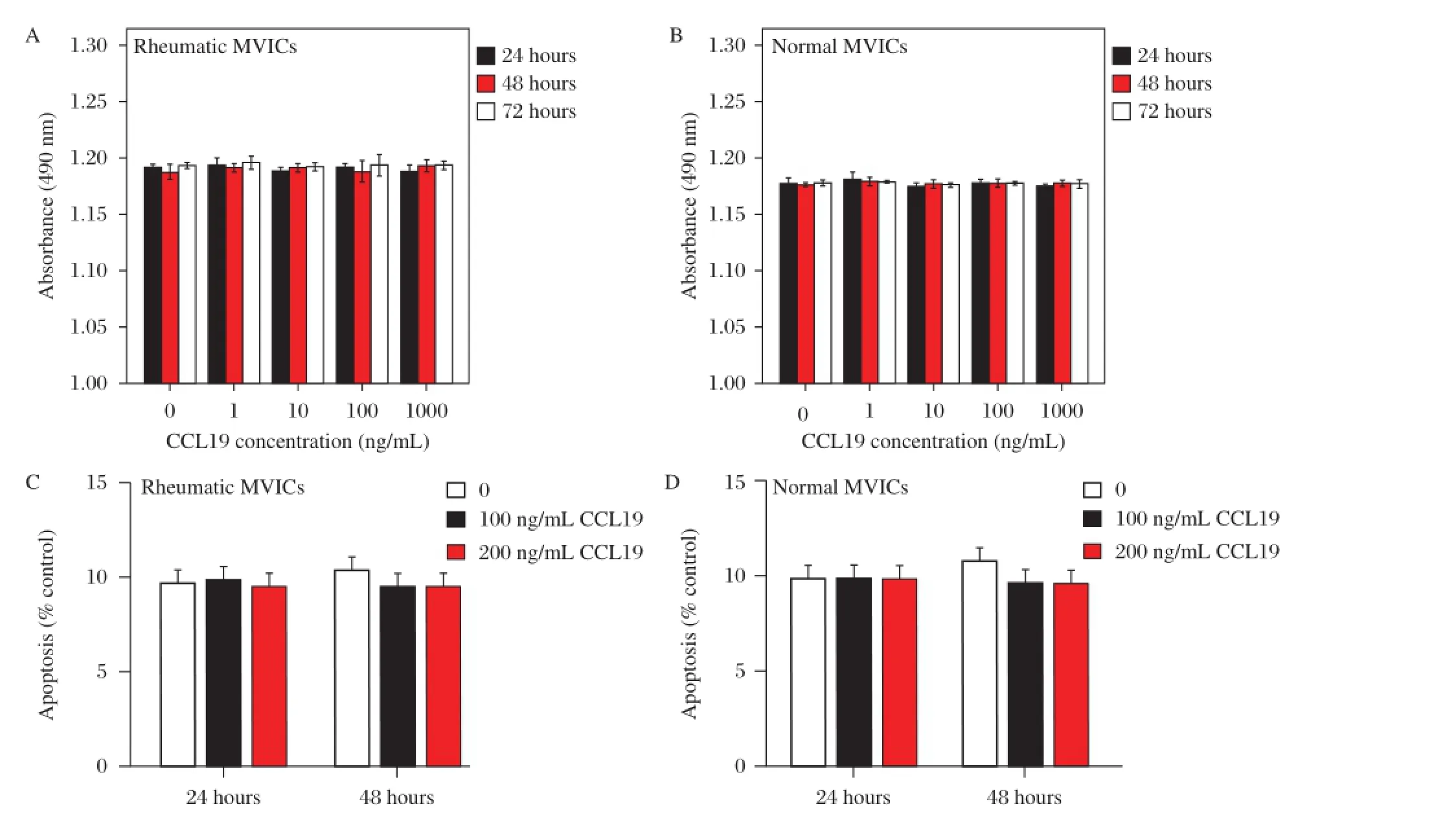
Fig.5CCL19 did not affect hMVICs proliferation or apoptosis.A-B)Both in the rheumatic and normalgroups,the proliferation of hMVICs showed no change in the presence of CCL19 from 1 ng/ml to 1000 ng/mlafter 24,48 and 72 h.C-D)No significantdifference was found at24h,48h after CCL19 treatment with respect to cell apoptosis both in rheumatic and normalgroups.
In this study,we demonstrated that CCR7 and CCL19 were expressed in human rheumatic stenotic mitral valves,but not in normal valves.CCR7 was mainly expressed in hMVICs in vivo,while mononuclear cells and the endothelium within the rheumatic mitral valves were the major source of CCL19. hMVICs treated with CCL19 showed enhanced wound healing rates,as a resultofenhanced migration.Taken together,our results indicated that CCR7 and CCL19 may be therapeutic targets to prevent mitral valve remodeling following rheumatic fever.
CCR7 and its ligand CCL19 play importantroles in lymphocyte homing to lymph nodes and balancing immunity and peripheraltolerance[6].Severalinvestigations have also shown that CCR7 is expressed by malignant cells involved in cancer metastasis[7,18-19]and by peripheral blood fibrocytes in response to wound healing[8,20].This indicates thatthe chemotactic effects of CCR7 can be exerted on non-inflammatory cells.CCL19,but not CCL21,effectively stimulates CCR7 phosphorylation and internalization[21],leading to receptor desensitization.This suggests that CCR7-mediated cellresponses to CCL19 may have a shorter time span,and CCL19-induced migration can be terminated[22].The understanding ofbiology and pathophysiology of valve interstitial cells(VICs)remains poor.Previous studies showed cultured VICs maintained theirmolecular phenotype up to eightpassages, itis accepted thatcultured VICs of3 to 5 passage were capable for in vitro study[15,23-24].In our study,CCR7 was highly expressed in cultured rheumatic hMVICs, butnotin normalhMVICs indicating the changed gene expression profiling of diseased MVICs.CCR7 was also not found in normal hMVICs in vivo,which may be due to the selection of relatively healthy valves. Interestingly,in cultured normalhMVICs,CCR7 was expressed significantly after treatment with CCL19. There has been no report of CCL19 directly promoting the expression of CCR7 in hMVICs.CCL19-induced up-regulation of CCR7 can be stimulated through NF-k B[18]or SOCS1[25].
Migration is an essential process in wound repair. Mesenchymal cells migrate into the wound from surrounding tissue and become activated to transform into myofibroblasts.hMVICs are involved in many aspects of wound healing,including migration,proliferation, apoptosis,and ECM remodeling[26].Injury to the valvular endothelium may initiate the cascade of wound repair events thatstimulate inflammation,eliciting a response in the underlying h MVICs to promote the accumulation of cells and ECM to promote valvular calcification[10,27].The release of growth factors and cytokines occurs immediately after injury.Tissuegrowth factor-β(TGF-β)and fibroblast growth factor-2(FGF-2)have been reported to regulate the migration and proliferation of valve interstitial cells following injury in vitro[15,28].
The role ofchemokines in valve repair has notbeen fully elucidated.In our study of the rheumatic mitral valves,CCR7 was mainly expressed in diseased hMVICs in vivo and in vitro.CCL19 and CCL21 are the sole ligands of CCR7.Therefore,secreted CCL19 expressed by mononuclear cells and endothelial cells may notonly contribute to the homing oflymphocytes and dendritic cells,butalso regulate the cellular behavior of CCR7-expressing h MVICs.In our study,the longer migration distance and accelerated closure of hMVICs from both the rheumatic and normal valves treated with CCL19 supported this hypothesis and suggesta novelrole ofthe CCR7/CCL19 axis in rheumatic valve remodeling.
In addition to CCR7,CCL19 also bindswith high affinity to the hepta-helicalsurface protein,termed the CCX-chemokine receptor(CCX-CKR)[29].Our findings demonstrated thatthe effectof CCL19 was completely abrogated by CCR7 neutralization,suggesting a dominantrole of the CCR7 pathway.Concentration dependentmigration was only observed fornormalhMVICs treated with CCL19,suggesting that h MVICs in the rheumatic valves were already stimulated.
In conclusion,our findings supportthe conceptthat activation of CCR7 by CCL19 regulates inflammatory cells,endothelialcells and hMVICs in rheumatic valve disease.Ourresults highlighta noveltissue remodeling mechanism mediated by the CCR7/CCL19 axis that may provide a clinical target for the treatment of chronic rheumatic mitral valve disease.
Acknowledgment
This work was supported by grants from the National NaturalScience Foundation ofChina(No.81100162)and the Priority Academic Program Developmentof Jiangsu Higher Education Institutions(PAPD2010-2013).
[1]Nkomo VT.Epidemiology and prevention of valvular heart diseases and infective endocarditis in Africa[J]. Heart 2007,93(13):1510-1519.
[2]Chopra P,Gulwani H.Pathology and pathogenesis of rheumatic heart disease[J].Indian J Pathol Microbiol 2007,50(4):685-697.
[3]Cunningham MW.Autoimmunity and molecular mimicry in the pathogenesis of post-streptococcal heart disease[J]. Front Biosci 2003,8(8):s533-543.
[4]Roberts S,Kosanke S,Terrence Dunn S,etal.Pathogenic mechanisms in rheumatic carditis:focus on valvular endothelium[J].J Infect Dis 2001,183(3):507-511.
[5]Guilherme L,Cury P,Demarchi LM,et al.Rheumatic heart disease:proinflammatory cytokines play a role in the progression and maintenance of valvular lesions[J]. Am J Pathol 2004,165(5):1583-1591.
[6]Forster R,Davalos-Misslitz AC,Rot A.CCR7 and its ligands:balancing immunity and tolerance[J].Nat Rev Immunol 2008,8(5):362-371.
[7]Muller A,Homey B,Soto H,etal.Involvementof chemokine receptors in breastcancermetastasis[J].Nature 2001, 410(6824):50-56.
[8]Abe R,Donnelly SC,Peng T,etal.Peripheralblood fibrocytes:differentiation pathway and migration to wound sites[J].J Immunol 2001,166(12):7556-7562.
[9]Kaur D,Saunders R,Berger P,etal.Airway smooth muscle and mast cell-derived CC chemokine ligand 19 mediate airway smooth muscle migration in asthma[J].Am J Respir Crit Care Med 2006,174(11):1179-1188.
[10]Durbin AD,Gotlieb AI.Advances towards understanding heart valve response to injury[J].Cardiovasc Pathol 2002,11(2):69-77.
[11]Liu AC,Joag VR,Gotlieb AI.The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology[J].Am J Pathol 2007,171(5):1407-1418.
[12]Chopra P,Tandon HD,Raizada V,etal.Comparative studies of mitral valves in rheumatic heart disease[J].Arch Intern Med 1983,143(4):661-666.
[13]Veinot JP.Pathology of inflammatory native valvular heart disease[J].Cardiovasc Pathol 2006,15(5):243-251.
[14]Taylor PM,Allen SP,Yacoub MH.Phenotypic and functional characterization of interstitial cells from human heart valves,pericardium and skin[J].J Heart Valve Dis 2000,9(1):150-158.
[15]Han L,Gotlieb AI.Fibroblastgrowth factor-2 promotes in vitro mitralvalve interstitialcellrepair through transforming growth factor-beta/Smad signaling[J].Am J Pathol 2011,178(1):119-127.
[16]Vermes I,Haanen C,Steffens-Nakken H,et al.A novel assay for apoptosis.Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V[J].J Immunol Methods 1995,184(1):39-51.
[17]Guidelines forthe diagnosis ofrheumatic fever.Jones Criteria, 1992 update.Special Writing Group of the Committee on Rheumatic Fever,Endocarditis,and Kawasaki Disease of the Council on Cardiovascular Disease in the Young of the American Heart Association[J].JAMA 1992,268(15):2069-2073.
[18]Mburu YK,Abe K,Ferris LK,etal.Human beta-defensin 3 promotes NF-kappaB-mediated CCR7 expression and anti-apoptotic signals in squamous cell carcinoma of the head and neck[J].Carcinogenesis 2011,32(2):168-174.
[19]Li P,Liu F,Sun L,etal.Chemokine receptor 7 promotes cell migration and adhesion in metastatic squamous cell carcinoma of the head and neck by activating integrin alphavbeta3[J].Int J Mol Med 2011,27(5):679-687.
[20]Mori L,Bellini A,Stacey MA,etal.Fibrocytes contribute to the myofibroblast population in wounded skin and originate from the bone marrow[J].Exp Cell Res 2005, 304(1):81-90.
[21]Kohout TA,Nicholas SL,Perry SJ,et al.Differential desensitization,receptor phosphorylation,beta-arrestin recruitment,and ERK1/2 activation by the two endogenousligands for the CC chemokine receptor 7[J].J Biol Chem 2004,279(22):23214-23222.
[22]Otero C,Groettrup M,Legler DF.Opposite fate of endocy tosed CCR7 and its ligan d s:recyclin g versus degradation[J].J Immunol 2006,177(4):2314-2323.
[23]Heaney AM,Bulmer BJ,Ross CR,et al.A technique for in vitro culture of canine valvular interstitialcells[J].J Vet Cardiol 2009,11(1):1-7.
[24]Connolly JM,Bakay MA,Fulmer JT,et al.Fenfluramine disrupts the mitral valve interstitial cell response to serotonin[J].Am J Pathol 2009,175(3):988-997.
[25]Yu CR,Mahdi RM,Liu X,et al.SOCS1 regulates CCR7 expression and migration of CD4+T cells into peripheral tissues[J].J Immunol 2008,181(2):1190-1198.
[26]Li C,Xu S,Gotlieb AI.The response to valve injury.A paradigm to understand the pathogenesis of heart valve disease[J].Cardiovasc Pathol 2011,20(3):183-190.
[27]Werner S,Grose R.Regulation of wound healing by growth factors and cytokines[J].Physiol Rev 2003,83(3): 835-870.
[28]Liu AC,Gotlieb AI.Transforming growth factor-beta regulates in vitro heart valve repair by activated valve interstitial cells[J].Am J Pathol 2008,173(5):1275-1285.
[29]Gosling J,Dairaghi DJ,Wang Y,et al.Cutting edge: identification of a novel chemokine receptor that binds dendritic cell-and T cell-active chemokines including ELC,SLC,and TECK[J].J Immunol 2000,164(6): 2851-2856.
△These authors contributed equally to this work.
✉Corresponding author:Dr.Xiangqing Kong,Department of Cardiology,The First Affiliated Hospital of Nanjing Medical University,No.300 Guangzhou Road,Nanjing,Jiangsu 210029, China.Tel/Fax:+86 25 83672050/+86 25 84352775,E-mail:xiangqing_kong@hotmail.com.
Received 25 February 2015,Revised 31 March 2015,Accepted 09 July 2015,Epub 09 September 2015
R329.2+5,Document code:A
The authors reported no conflict of interests.
©2015 by the Journal of Biomedical Research.All rights reserved.
10.7555/JBR.29.20150031
 THE JOURNAL OF BIOMEDICAL RESEARCH2015年6期
THE JOURNAL OF BIOMEDICAL RESEARCH2015年6期
- THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- Platelets in hemostasis and thrombosis:Novelmechanisms of fibrinogen-independent platelet aggregation and fibronectinmediated protein wave of hemostasis
- Retinolbinding protein 4 correlates with and is an early predictor of carotid atherosclerosis in type 2 diabetes mellitus patients
- Effectofburden and origin sites ofpremature ventricular contractions on left ventricular function by 7-day Holter monitor
- Prevention ofatrialfibrillation with renin-angiotensin system inhibitors on essentialhypertensive patients:a meta-analysis of randomized controlled trials
- Epirubicin-gold nanoparticles suppress hepatocellular carcinoma xenograft growth in nude mice
- A susceptibility locus rs7099208 is associated with non-obstructive azoospermia via reduction in the expression of FAM160B1