
母鼠营养不良导致子代在生命早期出现糖脂代谢紊乱及其机制探讨
2015-01-03 02:58郑佳肖新华张茜于淼许建萍王志新刘一静李明敏
遗传 2015年1期
郑佳,肖新华,张茜,于淼,许建萍,王志新,刘一静,李明敏
中国医学科学院,北京协和医学院,北京协和医院内分泌科,卫生部内分泌重点实验室,北京 100730
母鼠营养不良导致子代在生命早期出现糖脂代谢紊乱及其机制探讨
郑佳,肖新华,张茜,于淼,许建萍,王志新,刘一静,李明敏
中国医学科学院,北京协和医学院,北京协和医院内分泌科,卫生部内分泌重点实验室,北京 100730
为了探讨母鼠孕期和哺乳期营养不良对子代生命早期糖脂代谢的影响及其机制,文章对孕期和哺乳期母鼠分别喂养高脂饮食、低蛋白饮食和正常饮食,观察其子鼠断乳时(3周龄)糖脂代谢指标,并采用荧光定量PCR方法检测子鼠肝组织氧化物酶增殖物激活受体γ(PPARγ)基因的表达情况。结果表明:子鼠在3周龄时,与正常饮食组相比,低蛋白饮食组子鼠出生体重(7.36±0.91 vs 8.94±1.39,P<0.0001)较低,体长较短(12.27±0.53 vs 13.44±0.36,P<0.0001);高脂饮食组子鼠体重(9.53±0.68 vs 7.36±0.91,P<0.0001)和体长(13.22±0.35 vs 12.27±0.53,P<0.0001)均高于低蛋白饮食组;另外,高脂饮食组子鼠腹腔糖耐量实验30 min和60 min血糖明显高于正常饮食组(P<0.001),且高脂饮食组30 min血糖水平也明显高于低蛋白饮食组(P<0.001),高脂饮食组子鼠糖耐量曲线下面积明显大于正常饮食组(P<0.001)。另外,与正常饮食组相比,高脂饮食组子鼠空腹胆固醇水平明显升高(1.64±0.21 vs 1.18±0.16,P<0.01),低蛋白饮食组空腹胆固醇水平明显下降(0.96±0.09 vs 1.18±0.16,P<0.05)。荧光定量PCR结果显示,在低蛋白饮食组和高脂饮食组,其子鼠肝组织PPARγ基因表达量均明显高于正常饮食组(P<0.05)。结果显示,母鼠妊娠期和哺乳期高脂饮食与低蛋白饮食均可以诱导子鼠在发育早期出现糖脂代谢紊乱,PPARγ基因可能在其中参与了重要的调控作用。
母体营养;糖脂代谢;子鼠;PPARγ
研究发现,在妊娠后期处于饥饿环境下的母亲,低出生体重儿发生率高,并且这些低出生体重儿在成年期患心血管疾病、胰岛素抵抗、高血压的概率明显增加[1]。对动物的研究也同样表明,孕期营养缺乏会导致子代出现糖耐量下降和胰岛素抵抗[2]。因此英国学者提出了“胎儿代谢编程假说”,认为生命早期包括孕期和(或)哺乳期营养不良对子代有长远影响[3]。
近年来,随着孕妇肥胖和妊娠糖尿病发病率的不断增加,人们也越来越关注早期营养过剩对人类健康所带来的影响。母鼠营养过剩可以导致子鼠肥胖、糖耐量异常、胰岛素抵抗和血脂紊乱[4~6]。然而,这些研究普遍关注的是受宫内营养不良影响的子鼠在成年期甚至老年期的代谢影响,而子鼠生命早期如断乳时是否也存在代谢紊乱仍不清楚。
过氧化物酶增殖物激活受体(Peroxisome proliferator-activated receptors, PPARs)是核受体超家族成员之一,对组织重塑、炎症反应和血管形成等生物学过程发挥重要的调节作用。PPARs包括PPARα、PPARβ、PPARγ 3种亚型,其中PPARγ被认为是肥胖、胰岛素抵抗、糖尿病、代谢综合征等疾病防治的重要靶点[7]。目前,关于早期发育营养状况对子代生命早期糖脂代谢影响的机制仍不十分清楚,而PPARγ是否参与这一过程鲜有报道。
因此,本研究分别观察母鼠在营养不良和营养过剩两种不良的早期发育营养环境下,子鼠生命早期糖脂代谢的变化,并初步探讨PPARγ在这一过程中所起的调控作用。
1 材料和方法
1.1 实验动物、分组及处理
7周龄无特定病原体级别的C57BL/6J小鼠,购于北京维通利华实验动物技术有限公司,饲养于中国医学科学院医学实验动物研究所。3种饲料(正常饲料、低蛋白饲料和高脂饲料)均由北京科澳协力饲料有限公司提供。基于文献报道的实验动物模型[8,9],3种饲料的能量和质量组成见表1。
7周龄C57BL/6J雌性和雄性小鼠给予1周时间适应环境,期间均给予正常饮食。于 8周龄时进行交配:下午7时雌雄鼠2:1合笼,翌日上午6时观察,以观察到阴栓确定怀孕并记录为怀孕第 0.5 d。孕鼠单笼饲养并随机分为正常饮食组(Control,C)、低蛋白饮食组(Low protein, LP)和高脂饮食组(High fat, HF),具体实验流程如图1所示。饲养期间均自由摄食饮水,昼夜各12 h循环,环境温度保持在22±2℃,湿度适宜,每天记录摄食情况。

表1 3种不同饲料质量与能量组成比
子鼠于3周龄(断乳)时空腹12 h后处死,从眼眶静脉丛采血,4000 r/min、5 min常温离心后取血清,-80℃保存以进行代谢指标的测定。所有操作均在中国医学科学院医学实验动物研究所进行,均经过中国医学科学院医学实验动物研究所动物伦理委员会审核通过。

图1 动物实验流程图
1.2 方法
1.2.1 母鼠体重和血糖测量
母鼠在孕前进行体重和血糖的测量,在孕期和哺乳期每周测量1次体重。子鼠断乳时进行腹腔糖耐量实验,即禁食12 h后,取尾静脉血用血糖仪(德国拜安康)测定空腹血糖值,记为BG(Blood Glucose)0。随后,给予20%葡萄糖溶液(2 mg/g 体重)腹腔注射,分别于注射后30 min、60 min 和120 min取尾静脉血测定血糖值,记为BG30、BG60 及BG120。根据4个时间点的血糖值计算糖耐量曲线下面积(Area under the curve, AUC)。糖耐量曲线下面积公式为:AUC=0.5×(BG0+BG30)/2+0.5×(BG30+BG60)/2+1× (BG60+BG120)/2[10]。
1.2.2 子鼠体重和腹腔糖耐量实验
子鼠于出生第0.5 d记录出生体重,为了减少窝仔数量的差异,出生体重=窝重/只数,在哺乳期每只子鼠每3 d测量1次体重。在断乳时,用软尺测量子鼠从嘴尖至尾尖的距离,记录为体长。子鼠于3周龄(断乳)时进行腹腔糖耐量实验,方法同母鼠。
1.2.3 子鼠胰岛素测定和HOMA-IR值
子鼠血清胰岛素测定采用小鼠超敏酶联免疫吸附实验(ELISA),试剂盒购于美国 ALPCO公司(80-INSMSU-E01, Salem, NH),批间差异为4.2%。所有操作均严格按照试剂操作说明书进行,并且所有样本均进行双孔重复。另外,采用胰岛素抵抗指数稳态模型(HOMA-IR)评估子代小鼠胰岛素抵抗的情况。
HOMA-IR的计算公式为:HOMA-IR值=空腹胰岛素 (μU/mL) ×空腹血糖(mmol/L)/22.5[11]。
1.2.4 子鼠甘油三酯和胆固醇的测定
子鼠血清甘油三酯和胆固醇的测定采用比色法,试剂盒均购于美国BioVision公司。所有操作均严格按照试剂操作说明书进行,并且所有样本均进行双孔重复。
1.2.5 子鼠肝脏组织基因表达检测
1.2.5.1 肝脏组织总RNA提取及cDNA合成 将子鼠处死后,立即取肝脏组织置于干冰保存。用试剂盒提取总 RNA。用 Nanodrop (ND-1000) 检测RNA的浓度,并用琼脂糖凝胶电泳对RNA质量进行鉴定。取1 μg总RNA,用试剂盒(A3500, Promega)将总RNA逆转录成cDNA。
1.2.5.2 荧光定量PCR 利用荧光定量PCR方法检测PPARγ基因在正常饮食组、低蛋白饮食组以及高脂饮食组子鼠表达的差异。利用 OLIGO7设计PPARγ基因和β-actin内参基因扩增引物,序列如下:
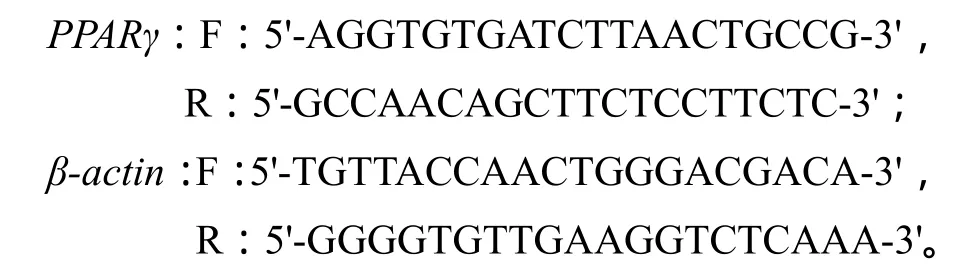
取 2 μL cDNA,配置成 20 μL反应体系,用SYBR® Green PCR Master Mix (RR420A, TaKaRa Bio Inc., Japan)试剂,使用ABI prism Vii7 Sequence Detection System(ABI Prism® Vii7, Applied Biosystems, Life Technologies) 仪器进行检测。荧光定量PCR扩增条件为:95℃预变性10 min;95℃变性10 s,55℃复性20 s,72℃延伸10 s,共进行40个循环。通过2-ΔΔCt计算正常饮食组、低蛋白饮食组以及高脂饮食组基因的表达情况。每个样本均重复3次实验。
1.2.6 统计学分析
采用SPSS15. 0 统计软件进行统计学分析,计量资料数据用均数±标准差表示,各组之间的比较采用方差分析,P<0.05 认为差异具有统计学意义。

图2 子鼠断乳时母鼠的体重、血糖情况
2 结果与分析
2.1 母鼠体重、血糖情况
母鼠孕前基线体重和血糖水平在 3组间均无明显差异。在子鼠断乳时,高脂饮食组和低蛋白饮食组母鼠分别与正常饮食组相比体重和血糖均无明显变化。高脂饮食组母鼠体重及糖耐量实验30 min和60 min的血糖明显高于低蛋白饮食组(P<0.05)(图2)。
2.2 子鼠体重与体长情况
正常饮食组、低蛋白饮食组及高脂饮食组子鼠间出生体重无明显差异。3周龄时,与正常饮食组相比,低蛋白饮食组子鼠出生体重较低(7.36±0.91 vs 8.94±1.39,P<0.001)和体长较短(12.27±0.53 vs 13.44±0.36,P<0.001),高脂饮食组与正常饮食组相比子鼠出生体重和体长无明显差异。高脂饮食组子鼠出生体重(9.53±0.68 vs 7.36±0.91,P<0.001)和体长(13.22±0.35 vs 12.27±0.53,P<0.001)均高于低蛋白饮食组(表2)。3周龄时,高脂饮食组与正常饮食组的雄性和雌性子鼠体重均无统计学差异(P>0.05)。而低蛋白饮食组雄性和雌性子鼠体重均低于正常饮食组和高脂饮食组(P<0.05)(图3)。
2.3 子鼠3周龄糖耐量情况和曲线下面积
3周龄子鼠空腹血糖在正常饮食组、低蛋白饮食组以及高脂饮食组间无明显差异(表3)。高脂饮食组子鼠经腹腔糖耐量实验,30 min和60 min血糖明显高于正常饮食组(P<0.001),高脂饮食组与低蛋白饮食组相比,30 min血糖明显升高(P<0.001)。高脂饮食组雄性子鼠经腹腔糖耐量实验,30 min(P<0.001) 和60 min(P<0.01)血糖明显高于正常饮食组。

表2 子鼠出生体重和3周龄体重与体长的比较

图3 3组雌、雄子鼠3周龄时体重的比较
低蛋白饮食组雄性子鼠0 min血糖高于正常饮食组(P<0.05),与低蛋白饮食组相比,高脂饮食组雄性子鼠30 min血糖明显升高(P<0.001)。高脂饮食组雌性子鼠经腹腔糖耐量实验,30 min(P<0.01)血糖高于正常饮食组。与低蛋白饮食组相比,高脂饮食组雌性子鼠30 min血糖明显升高(P<0.001)(图4)。高脂饮食组糖耐量曲线下面积明显大于正常饮食组(P<0.001),而高脂饮食组与低蛋白饮食组比较无差异(表3)。
2.4 子鼠3周龄血清胰岛素和胰岛素抵抗指标的评价
3周龄时,正常饮食组、低蛋白饮食组及高脂饮食组子鼠空腹胰岛素水平和 HOMA-IR值均无明显统计学差异(P>0.05) (表3)。

图4 3组子鼠3周龄腹腔糖耐量情况

表3 3组子鼠3周龄时各项代谢相关指标的比较
2.5 子鼠3周龄血脂情况
3周龄时,正常饮食组、低蛋白饮食组以及高脂饮食组子鼠空腹甘油三酯水平无统计学差异(P>0.05)。高脂饮食组子鼠空腹胆固醇水平较正常饮食组和低蛋白饮食组均明显升高(1.64±0.21 vs 1.18±0.16,P< 0.01;1.64±0.21 vs 0.96±0.09,P<0.001),与正常饮食组相比,低蛋白饮食组子鼠空腹胆固醇水平下降(0.96± 0.09 vs 1.18±0.16,P<0.05)(表3)。3组雄性和雌性子鼠空腹甘油三酯水平均无统计学差异(P>0.05)。雄性和雌性子鼠空腹胆固醇水平高脂饮食组均高于正常饮食组和低蛋白饮食组,正常饮食组高于低蛋白饮食组,差异均具有统计学意义(P<0.05)(图5)。
2.6 子鼠3周龄PPARγ基因表达情况
荧光定量PCR结果显示:PPARγ在高脂饮食组、低蛋白饮食组和正常饮食组子鼠肝脏组织均有较高的表达。PPARγ基因在低蛋白饮食组和高脂饮食组较正常饮食组表达量均升高,相对表达量分别为1.4±0.17和1.56±0.12(P<0.05)。而高脂饮食组与低蛋白相比无明显统计学差异(P>0.05),雄性和雌性表达无明显差异(图6)。

图5 3组雌雄子鼠3周龄时血清胆固醇的比较

图6 3组子鼠3周龄时PPARγ基因表达情况比较
3 讨 论
目前普遍认为遗传与环境因素相互作用是2型糖尿病、心血管疾病和肥胖病等代谢性疾病发生发展的主要机制。近年来,除了传统的环境因素,如饮食、运动、肥胖外,人们越来越关注生命早期发育环境(包括妊娠期和哺乳期)对成年期慢性疾病的影响。大量的临床研究和动物实验表明,早期营养不良者(包括营养缺乏与营养过剩)在成年期患肥胖、动脉硬化、胰岛素抵抗、糖尿病等代谢性疾病的风险明显升高[12~17]。
本研究进一步观察发现,孕期和哺乳期母鼠在高脂饮食和低蛋白饮食下,其子鼠在生命早期即断乳时,即已出现糖耐量下降,而且,高脂饮食组的子鼠血清胆固醇水平也明显升高。既往有研究显示,母鼠在孕前、孕期和哺乳期持续高脂饮食能够导致子代在8周至36周龄出现代谢性疾病,如肥胖、血脂紊乱、胰岛素抵抗和糖尿病[18~20]。另外有研究显示,孕期的低蛋白饮食能够导致子代在72周龄出现高甘油三酯血症和胰岛素抵抗[3]。然而,这些动物模型都局限于对子鼠成年期甚至老年期的影响,并不清楚这种不良的影响是何时开始出现的。因此与以往研究不同的是,本研究关注的是早期营养不良(包括营养缺乏与营养过剩)对子代生命早期(断乳时)的影响,结果提示,在断乳时这种不良的影响就已经存在。
另外,本研究显示,高脂饮食组子鼠糖耐量实验30 min血糖、糖耐量曲线下面积和空腹胆固醇水平均高于低蛋白饮食组,这表明高脂饮食所带来的糖脂代谢紊乱的易感性可能会大于低蛋白饮食。母鼠在孕期和哺乳期的低蛋白饮食和高脂饮食导致子代出现不同程度的糖脂代谢的异常,这主要是由于母鼠摄入能量的差异造成的,既往大量研究证实高脂饮食和低蛋白饮食分别属于能量过剩和能量缺乏的营养状况[8,9]。妊娠期和哺乳期是个体发育的关键时期,近年来,随着生活水平的提高,孕妇肥胖和妊娠糖尿病发病率的显著增加可能是导致子代发生糖脂代谢紊乱的原因。因此,人们对妊娠期和哺乳期进行积极有效的饮食和运动干预可能会在一定程度上改善甚至逆转子代糖脂代谢的紊乱。
本研究还发现,低蛋白饮食组和高脂饮食组PPARγ基因表达量均高于正常饮食组,这一结果与已往报道的研究相一致。既往研究显示,母体营养不良导致子代在断乳时出现肥胖,并且PPARγ表达增加[21]。PPARγ+/-杂合突变小鼠的胰岛素敏感性高于野生正常表达PPARγ水平的小鼠,显示内源性配体过度激活PPARγ与胰岛素抵抗和糖尿病的发生正相关[22]。另外,动物模型和临床试验显示外源性的PPARγ调节剂如小檗碱具有改善胰岛素敏感性的作用,这提示PPARγ调节剂可能是潜在的药物作用靶点[23]。
综上所述,母鼠妊娠期和哺乳期营养缺乏与营养过剩均导致子代在生命早期出现糖脂代谢紊乱。可能是因为母体不良的营养状况使子代糖脂代谢相关的基因在生命早期表达发生了改变[20]。此外,对PPARγ的进一步研究可能为预防和控制生命早期的糖脂代谢异常提供理论基础。
[1]Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet, 1989, 2(8663): 577-580.
[2]Erhuma A, Salter AM, Sculley DV, Langley-Evans SC, Bennett AJ. Prenatal exposure to a low-protein diet programs disordered regulation of lipid metabolism in the aging rat. Am J Physiol Endocrinol Metab, 2007, 292(6): E1702-E1714.
[3]Lucas A, Fewtrell MS, Cole TJ. Fetal origins of adult disease-the hypothesis revisited. BMJ, 1999, 319(7204): 245-249.
[4]Treesukosol Y, Sun B, Moghadam AA, Liang NC, Tamashiro KL, Moran TH. Maternal high-fat diet during pregnancy and lactation reduces the appetitive behavioral component in female offspring tested in a brief-access taste procedure. Am J Physiol Regul Integr Comp Physiol, 2014, 306(7): R499-R509.
[5]Zhang JL, Zhang F, Didelot X, Bruce KD, Cagampang FR, Vatish M, Hanson M, Lehnert H, Ceriello A, Byrne CD. Maternal high fat diet during pregnancy and lactation alters hepatic expression of insulin like growth factor-2 and key microRNAs in the adult offspring. BMC Genomics, 2009, 10: 478.
[6]Yokomizo H, Inoguchi T, Sonoda N, Sakaki Y, Maeda Y, Inoue T, Hirata E, Takei R, Ikeda N, Fujii M, Fukuda K, Sasaki H. Maternal high-fat diet induces insulin resistance and deterioration of pancreatic beta-cell function in adult offspring with sex differences in mice. Am J Physiol En-docrinol Metab, 2014, 306(10): E1163-E1175.
[7]Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, Evans RM. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med, 2013, 19(5): 557-566.
[8]Lillycrop KA, Phillips ES, Jackson AA, Hanson MA, Burdge GC. Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J Nutr, 2005, 135(6): 1382-1386.
[9]Winzell MS, Ahrén B. The high-fat diet-fed mouse: a model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes, 2004, 53 (Suppl.3): S215-S219.
[10]Purves RD. Optimum numerical integration methods for estimation of area-under-the-curve (AUC) and area-underthe-moment-curve (AUMC). J Pharmacokinet Biopharm, 1992, 20(3): 211-226.
[11]Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia, 1985, 28(7): 412-419.
[12]McKay JA, Xie L, Manus C, Langie SA, Maxwell RJ, Ford D, Mathers JC. Metabolic effects of a high-fat diet post-weaning after low maternal dietary folate during pregnancy and lactation. Mol Nutr Food Res, 2014, 58(5): 1087-1097.
[13]Watkins AJ, Sinclair KD. Paternal low protein diet affects adult offspring cardiovascular and metabolic function in mice. Am J Physiol Heart Circ Physiol, 2014, 306(10): H1444-H1452.
[14]Goyal R, Longo LD. Maternal protein deprivation: sexually dimorphic programming of hypertension in the mouse. Hypertens Res, 2013, 36(1): 29-35.
[15]Benatti RO, Melo AM, Borges FO, Ignacio-Souza LM, Simino LA, Milanski M, Velloso LA, Torsoni MA, Torsoni AS. Maternal high-fat diet consumption modulates hepatic lipid metabolism and microRNA-122 (miR-122) and microRNA-370 (miR-370) expression in offspring. Br J Nutr, 2014, 111(12): 2112-2122.
[16]Xiao XH, Zhang ZX, Cohen HJ, Wang H, Li WH, Wang T, Xu T, Liu AM, Gai MY, Ying S, Schmitz O, Yi Z. Evidence of a relationship between infant birth weight and later diabetes and impaired glucose regulation in a Chinese population. Diabetes Care, 2008, 31(3): 483-487.
[17]Xiao XH, Zhang ZX, Li WH, Feng K, Sun Q, Cohen HJ, Xu T, Wang H, Liu AM, Gong XM, Shen Y, Yi Z. Low birth weight is associated with components of the metabolic syndrome. Metabolism, 2010, 59(9): 1282-1286.
[18]Elahi MM, Cagampang FR, Mukhtar D, Anthony FW, Ohri SK, Hanson MA. Long-term maternal high-fat feeding from weaning through pregnancy and lactation predisposes offspring to hypertension, raised plasma lipids and fatty liver in mice. Br J Nutr, 2009, 102(4): 514-519.
[19]Borengasser SJ, Kang P, Faske J, Gomez-Acevedo H, Blackburn ML, Badger TM, Shankar K. High fat diet and in utero exposure to maternal obesity disrupts circadian rhythm and leads to metabolic programming of liver in rat offspring. PLoS One, 2014, 9(1): e84209.
[20]Masuyama H, Hiramatsu Y. Effects of a high-fat diet exposure in utero on the metabolic syndrome-like phenomenon in mouse offspring through epigenetic changes in adipocytokine gene expression. Endocrinology, 2012, 153(6): 2823-2830.
[21]Hyatt MA, Gardner DS, Sebert S, Wilson V, Davidson N, Nigmatullina Y, Chan LL, Budge H, Symonds ME. Suboptimal maternal nutrition, during early fetal liver development, promotes lipid accumulation in the liver of obese offspring. Reproduction, 2011, 141 (1): 119-126.
[22]Miles PD, Barak Y, He W, Evans RM, Olefsky JM. Improved insulin-sensitivity in mice heterozygous for PPAR-γ deficiency. J Clin Invest, 2000, 105(3): 287-292.
[23]Magliano DC, Bargut TCL, de Carvalho SN, Aguila MB, Mandarim-de-Lacerda CA, Souza-Mello V. Peroxisome proliferator-activated receptors-α and γ are targets to treat offspring from maternal diet-induced obesity in mice. PLoS One, 2013, 8(5): e64258.
(责任编委: 陈雁)
PPARγ links maternal malnutrition and abnormal glucose and lipid metabolism in the offspring of mice
Jia Zheng, Xinhua Xiao, Qian Zhang, Miao Yu, Jianping Xu, Zhixin Wang, Yijing Liu, Mingmin Li
Key Laboratory of Endocrinology of Ministry of Health; Department of Endocrinology, Peking Union Medical College Hospital; Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100730, China
Peroxisome proliferator-activated receptors (PPARs) are a group of nuclear receptor proteins that regulate gene transcription. PPARs play essential roles in modulating cell differentiation, development, and metabolism (carbohydrate, lipid, protein). Here, we investigated whether PPARγ plays a role in linking maternal malnutrition andaberrant metabolism in the offspring of mice. After feeding dams with high fat (HF) and low protein (LP) diet during pregnancy and lactation, we examined the effects on the offspring at weaning (age of 3-week). The results showed that the LP offspring had lower body weight and length than the control. The HF offspring had heavier body weight and longer body length than LP. The blood glucose levels in HF group were significantly higher at 30 min and 60 min after intraperitoneal glucose administration and the area under curve was also significantly larger than the control. The blood glucose levels in HF group were significantly higher at 30 min than LP. HF group had elevated total cholesterol levels and LP group had decreased total cholesterol levels compared with the control. All results were statistically significant as examined by t-test. More importantly, PPARγ expression levels detected by qRT-PCR were significantly increased in HF and LP groups compared with the control. In conclusion, maternal HF and LP diet during pregnancy and lactation can induce impaired glucose and lipid metabolism in the early life of mouse offspring, where PPARγ may play an important role.
maternal nutrition; glucose and lipid metabolism; mice; PPARγ
2014-05-10;
2014-08-07
国家自然科学基金项目(编号:81170736)资助
郑佳,在读博士研究生,研究方向:糖尿病发病机制的表观遗传学研究。E-mail: zhengjiapumc@163.com
肖新华,教授,博士生导师,研究方向:糖尿病发病机制。E-mail: xiaoxinhua@medmail.com.cn
10.16288/j.yczz.2015.01.010
时间: 2014-11-5 9:32:11
URL: http://www.cnki.net/kcms/detail/11.1913.R.20141105.0932.001.html
猜你喜欢
中国典型病例大全(2022年9期)2022-04-19
意林·少年版(2022年2期)2022-03-09
晚晴(2021年1期)2021-05-17
草堂(2020年1期)2020-02-18
新民周刊(2020年3期)2020-02-14
中国动物保健(2019年3期)2019-12-11
乐活老年(2018年8期)2018-11-26
鞍山师范学院学报(2018年4期)2018-10-23
医药前沿(2018年36期)2018-01-17
中国循环杂志(2015年10期)2015-12-24
