
识别杜洛克猪个体身份的DNA 条形码
2014-12-23 11:30邢光东胡肄农纪红军赵庆顺徐银学
江苏农业学报 2014年5期
丁 潜, 邢光东, 胡肄农, 纪红军, 赵庆顺, 徐银学
(1.南京农业大学动物科技学院,江苏 南京210095;2.江苏省农业科学院畜牧研究所,江苏 南京210014;3.江苏省农业科学院兽医研究所,江苏 南京210014;4.南京大学模式动物研究所,江苏 南京210061)
猪个体身份识别在猪场管理、猪肉产品溯源等方面意义重要。技术简单、成本低廉且操作简单的耳标等传统方法仍是猪场的主要个体识别方法,但应用中存在易损、易脱落丢失、易混淆等缺陷。基于传统方法的不足,近年来DNA 个体鉴定方法,包括微卫星标记、SNP 标记等在内的个体鉴定方法成为研究热点[1-3],但仍无法得到实际应用。针对以上情况,我们依据NCBI 数据库中已登录的SNP 相关信息[4],设计并筛选可有效且特异进行PCR 扩增的引物对,以获得高杂合度SNP 位点,用于猪个体身份DNA 条形码及相应的数字条形码编制,实现猪个体身份的DNA 识别。
1 材料与方法
1.1 试验材料及基因组DNA 模板制备
68 头杜洛克(Duroc)猪的耳组织样采自杭州大观山种猪场,-20 ℃冷冻保存备用。基因组DNA模板用DNA Mini Extract 试剂盒(南京润邦生物技术有限公司产品)制备,具体制备过程如下(单个样本):小米粒大小(1 mm3左右)耳组织放入装有17.6 μl DNA Mini Extract (A、B 液混合而成)的PCR 管中,PCR 仪中95 ℃5 min,16 ℃1 min(在此过程中暂停PCR 仪,加入10 mg/ml蛋白酶K 2.4 μl),55 ℃2 h,95 ℃10 min,16 ℃1 min,离心取上清或上清混合成模板作PCR 模板。
1.2 引物设计、PCR 反应、测序
根据NCBI 登录的猪基因组序列,共设计31 对引物,其中8 对引物的混合样本PCR 产物电泳条带单一,直接测序峰图背景干净易读且包含1 个或多个套峰,并获得H≥0.10 的SNP 位点(表1、表2)。引物扩增的PCR 体系(20 μl)为:样品基因组DNA模板1 μl,正反引物各1 μl,超纯水7 μl,2 × PCR Master Mix 10 μl。反应条件为:95 ℃先变性2 min;95 ℃变性30 s,退火30 s,72 ℃延伸50 s,共35 个循环;72 ℃最后延伸10 min。各引物对的退火温度和延伸时间见表1。PCR 扩增产物测序结果用Vector NTI Advance 11 软件比对,并结合分析测序峰图,以筛选SNP 位点。

表1 筛选的8 对引物相关信息及其退火温度和延伸时间Table 1 Information of 8 primer pairs and their annealing temperatures and extending times for PCR reactions
1.3 SNP 连锁不平衡分析和杂合度计算
基因的同一条染色体或扩增片段SNP 位点之间存在关联性,即SNP 位点连锁不平衡(Linkage disequilibrium,LD)现象。根据68 个杜洛克个体的扩增片段测序结果,对获得的39 个SNP 位点的关联性进行分析。剔除完全关联的SNP 位点,并过滤杂合度小于0.10 的SNP 位点,根据剩余的16 个SNP 位点,对扩增片段进行基因分型。
杂合度反映一个位点具有2 个以上等位基因的期望值。杂合度小的SNP 位点出现单一纯合基因型的概率较大,用于个体识别的意义也相对较小。为了使研发的DNA 个体身份识别技术更有效,选用杂合度H≥0.10 的SNP 位点。杂合度值的计算公式如下:
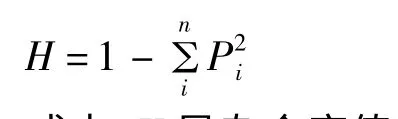
式中,H是杂合度值,Pi是每个SNP 的等位基因频率,n是等位基因的数目。
1.4 猪个体身份识别的SNP 条形码及相对应的数字条形码编制
8 对引物扩增的有效SNP 位点共计39 个,经连锁不平衡分析及杂合度计算,最终确定16 个SNP位点用于杜洛克猪个体身份识别的条形码编制。SNP 条形码中SNP 位点的排列,以引物对1 ~8 的次序依次排列(其中每对引物的SNP 位点以5'-至3'-顺序依次排列),组合成SNP 条形码。其中SNP位点以每对引物扩增产物(正向引物)的第1 个碱基所在位置计为+1 进行标识。每个SNP 位点可形成AA、TT、GG、CC、AT、AG、AC、TG、TC、GC 10 种基因型,当依次分别用数字0、1、2、3、4、5、6、7、8、9 共10 个数字替代时,每个个体不同SNP 位点的基因型就转换为相应的数字SNP 条形码。
2 结果与分析
2.1 SNP 杂合度筛选及连锁不平衡分析
8 对引物扩增68 头杜洛克猪基因组DNA 样并测序,共获得39 个SNP 位点(表2)。经SNP 位点连锁不平衡分析及杂合度筛选(H≥0.10),最终选留16 个SNP 位点用于杜洛克猪的个体身份识别(表2)。剔除的23 个SNP 位点,或与选留的SNP位点完全连锁,或其杂合度H<0.10。虽然杂合度H<0.10 的SNP 位点也能够与其他SNP 位点一起组成数量极少的基因型,但是因为杂合度过低,在繁育过程中容易产生单一纯合子而在后代中丢失多态性,在个体识别中意义不大。

表2 8 对引物在杜洛克猪中获得的39 个SNP 位点Table 2 The 39 SNP loci obtained by 8 PCR primer pairs in Duroc pigs
2.2 SNP 条形码及数字条形码编制
选留的16 个H≥0.10 的SNP 位点用于杜洛克猪个体身份识别条形码编制。在猪个体识别中,同窝猪样品的个体识别理论上是最困难的,试验中能够完全区分的68 个样品来自10 窝猪,而由这16 个SNP 组成的基因型能够在68 头杜洛克猪中,完全区分每个个体,实现个体识别(表3)。

表3 编制的用于个体身份识别的部分杜洛克猪SNP 条形码及相应的数字条形码Table 3 SNP and their corresponding digital barcodes of some of the Duroc pigs used for individual identification
3 讨论
3.1 猪个体身份识别
传统的耳标等猪个体识别技术[5],一旦与肉产品分离就会失去作用,而基因标记技术可以弥补这一缺陷[6]。基因标记技术将成为新一代的溯源或个体识别标记技术。在基因标记技术中,SNP 标记被公认为最具有潜在的实际应用价值。猪共有18对常染色体,每个SNP 位点存在3 种基因型,如果每对染色体仅筛选、利用1 个杂合度高的SNP 位点,那么理论上18 个SNP 位点就可以组成近4 亿(318=387 420 489)种基因型。实际上每条染色体上可利用的SNP 位点很多(1 对引物可同时扩增出多个SNP 位点,且可组成多种可利用的基因型),筛选SNP 位点并用于猪的个体身份识别是可行的。本研究筛选的8 对引物扩增片段中组成的基因型分别有2、10、9、3、3、9、3、3 种,理论上可以用于131 220(2 ×10 ×9 ×3 ×3 ×9 ×3 ×3)头猪的个体身份识别,试验中也很好地区分了所采集的68 头猪的样品。
3.2 混样PCR 扩增筛选SNP 位点
在NCBI 公共数据库中获得的候选SNP 被用到遗传学研究中之前,这些SNP 的验证和其等位基因频率的估计是必须的[7]。而对这些SNP 的等位基因频率估算,需要大量且相关度低的样品,成本很高[8]。通过混合等量个体DNA 样品形成混样来检测等位基因频率,高效且廉价,能够大大地减少DNA 的用量及分析费用[7]。这是本试验选择使用混合模板的原因。
我们先通过混样PCR 产物测序,粗略检测SNP位点存在情况,再由每个个体的SNP 位点检测,最终确定1 对引物所扩增的片段是否含有多个SNP位点存在。混样PCR 产物测序时会出现套峰,无论是个别优势模板PCR 的结果,或是部分模板PCR产物混合的结果,都能在一定程度上说明套峰出现位置存在SNP 位点。在混样测序的结果中,有3 处套峰都说明该处存在SNP,+502 处虽然在单个样品测序中发现了SNP,但在混合样品测序中并没有发现,查找其对应的杂合度可知,+502 处的杂合度为0.13,较其他3 处0.37 低了很多。说明混样测序中套峰的出现,在一定程度上表明该处存在SNP 且杂合度较高。
3.3 SNP 位点的连锁不平衡分析及杂合度筛选
SNP 位点连锁不平衡,是指同一条染色体上,2个SNP 位点间的非随机相关,即位于同一条染色体的2 个SNP 位点同时存在的概率,大于群体中因随机分布而同时出现的概率。2 个SNP 位点完全连锁不平衡时,这2 个SNP 位点组成的单体型只有2种,在用作个体身份识别时2 个一起的效果和单独一个是相同的,这样的SNP 位点只取其一即可。本试验中引物对3 共获得9 个SNP 位点,其中+470、+499 和+530 这3 个SNP 所组成的基因型只有TTTTTT、CCGGCC 和TCTGTC 这3 种,在其余65 个样品中这3 个SNP 也只能组成该3 种基因型。说明在该群体中,若排除新的SNP 出现,则+470、+499 和+530 这3 个SNP 只有TTT 和CGC 这2 种单体型,SNP 完全关联,所以+470、+499 和+530 这3 个SNP 中取+470 一个用作个体鉴定即可。而+502 位置和+ 470 位置的SNP 共可组成TCCC、TCTC、TTCC、CCCC 4 种基因型,推断这2 个位置有TC、CC、TT 这3 种单体型,SNP 没有完全关联,所以+470 和+502 都可用于个体识别。8 对引物扩增的SNP 位点经过连锁不平衡分析,完全连锁的SNP位点都只保留一个。其中,虽然引物对2 和引物对3 在同一条染色体上,但这2 对引物所扩增的片段在染色体上相距约9 Mbp,扩增的SNP 位点之间没有发现完全的连锁不平衡。杂合度过低(H<0.10)的SNP 位点对于个体识别的意义不大,编码SNP 条形码和对应的数字条形码时,予以舍弃。
[1] INAGAKI S,YAMAMOTO Y,DOI Y,et al.A new 39-plex analysis method for SNPs including 15 blood group loci[J].Forensic Sci Int,2004,144(1):45-57.
[2] LEE H Y,PARK M J,YOO J E,et al.Selection of twenty-four highly informative SNP markers for human identification and paternity analysis in Koreans[J].Forensic Sci Int,2005,148(2-3):107-112.
[3] ZENG Z,WANG L,FENG Q,et al.Evaluation of 96 SNPs in 14 populations for worldwide individual identification[J].Forensic Sci,2012,57(4):1031-1035.
[4] VAN TASSELL C P,SMITH T P,MATUKUMALLI L K,et al.SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries[J].Nat Methods,2008,5(3):247-252.
[5] MADEC F,GEERS R,VESSEUR P,et al.Traceability in the pig production chain[J].Rev Sci Tech,2001,20(2):523-537.
[6] GOFFAUX F,CHINA B,DAMS L,et al.Development of a genetic traceability test in pig based on single nucleotide polymorphism detection[J].Forensic Sci Int,2005,151(2-3):239-247.
[7] SASAKI T,TAHIRA T,SUZUKI A,et al.Precise estimation of allele frequencies of single-nucleotide polymorphisms by a quantitative SSCP analysis of pooled DNA[J].Hum Genet,2001,68(1):214-218.
[8] RISCH N,TENG J.The relative power of family-based and casecontrol designs for linkage disequilibrium studies of complex human diseases I.DNA pooling[J].Genome Res,1998,8(12):1273-1288.
猜你喜欢
少年文艺·开心阅读作文(2021年8期)2021-09-05
种子(2021年3期)2021-04-12
猪业科学(2020年5期)2020-12-11
猪业科学(2020年4期)2020-05-11
小学科学(学生版)(2019年5期)2019-05-21
少儿美术(快乐历史地理)(2019年11期)2019-04-20
猪业科学(2018年5期)2018-07-17
小学生导刊(2017年13期)2017-06-15
外语教学理论与实践(2016年1期)2016-06-11
科学种养(2014年10期)2014-10-23
