
Theoretical Study on the Structures and Properties of Phenobarbital Imprinted Polymers①
2014-12-18 01:24SUTingTingLIUJunBoTANGShnShnCHANGHiBoJINRui
结构化学 2014年10期
SU Ting-Ting LIU Jun-Bo② TANG Shn-Shn② CHANG Hi-Bo JIN Rui-F
Theoretical Study on the Structures and Properties of Phenobarbital Imprinted Polymers①
SU Ting-TingaLIU Jun-Boa②TANG Shan-Shana②CHANG Hai-BoaJIN Rui-Fab
a(130118)b(024000)
Recently, the investigation of novel molecularly imprinted polymers (MIPs) has attracted a lot of interest and becomes a fascinating field. The phenobarbital (PHN) was taken as an imprinted molecule and the 2-vinyl-4,6-diamino-1,3,5-triazine (VDAT) was considered as a functional monomer in this study. The geometry optimization, natural bond orbital (NBO) charge, and molecular electrostatic potential (MEP) of PHN and VDAT were studied at the M062X level belonging to one of the hybrid density functional theories. Furthermore, we discussed the bonding conditions of PHN molecular imprinted polymers (PHN-MIPs) via the hydrogen bond length and atoms in molecules (AIM) theory. The rebinding property of PHN-MIPs was also researched.The results of MEP and NBO charge analysis were coincident. The stability property was excellent when the ratio of PHN and VDAT was 1:4. Except the classic hydrogen bonds, non-classical hydrogen bonds also existed in the imprinted polymers. By simulating the rebinding energies between the pentobarbital (PNT), barbital (BAR), and PHN-MIPs after the elution of PHN,the rebinding property of PHN-MIPs to PHN was excellent when PNT and BAR existed all at once. This research can provide theoretical reference for the synthesis and characterization of novel PHN-MIPs.
phenobarbital, 2-vinyl-4,6-diamino-1,3,5-triazine, molecular imprinting, computer simulation, hydrogen bonds
1 INTRODUCTION
Molecularly imprinted polymers (MIPs) can iden- tify a substance specifically. Its principle is that MIPs contain a cavity afterthe elution of template molecules. The cavity is highly in accordance with the template molecules in respect of size, shape, and functional groups. Thus the MIPs could bond with the template molecules particularly. This principle makes the molecularly imprinted technology attract lots of concerns in many fields and the MIPs be con- sidered as recognition elements for a certain analyte, such as chromatographic separation[1], chemical sen- sors[2], solid phase extraction[3],and drug separa- tion[4].
The selective recognition of MIPs is affected by many factors, such as the functional monomer, im- printing ratio, porogen, and cross-linking agent. As the selective adsorption of MIPs depends mainly on the binding affinity between template molecules andfunctional monomers, the functional monomers de- termine directly the selective adsorption of polymers toward the template molecules. Recently, many researchers have applied the molecular simulation to the molecular imprinting systems to improve the selectivity and adsorption of MIPs[5−7].
At present, there are many documents about the extraction of triazine compounds via molecularly imprinted materials. For example, Djozan[8]synthe- sized the triazine-MIPs and these polymers could extract triazine herbicides (atrazine, simazine, propazine, terbutryn,.) obviously. Its extraction value was 88 ng/mL. Rongning Liang[9]synthesized the potentiometric sensor by using melamine as a template molecule, and this sensor has been success- fully applied to determine the melamine in milk samples. Liu Junbo[10]researched the interaction mechanism of atrazine-MIPs and this molecule be- longed to a triazine compound. Furthermore, atra- zine had the strongest interaction with trifluorome- thylacrylic acid. The literatures suggested that tria- zine compounds had fine imprinted binding capacity, but triazine compounds were concerned mostly as template molecules. Based on the imprinting effect of triazine compounds, we simulated these processes of molecular imprinting assembling, elution of imprinted molecules, and assembling once more via the computer simulation software. The whole pro- cesses used PHN as the template molecule and VDAT as the functional monomer at the M062X level. The purpose is to research the mechanism of interaction between the imprinted molecules and functional monomers and acquire the imprinting ratios. This research can provide a theoretical basis for the synthesis of novel MIPs materials and the recognition experiments.
2 COMPUTATINAL METHODS
In the field of molecular simulation, the theory of quantum mechanics calculation includingmethod, density functional theory (DFT), semi- empirical method, and perturbation theory can calcu- late the basic properties of molecules. The contribu- tion of electrons is important for molecular proper- ties, thus electron interaction can not be ignored[11]. Therefore, we chose DFT as the resourceful theory[12]. It is well-known that the accuracy and reliability calculated via different functional theories are different in the matter of molecular calculation. For example, the common B3LYP method has a good effect on the geometrical structure of mole- cules, but it admits flaws in certain hydrogen bond calculation[13, 14]. Thereby, we optimized the struc- ture of PHN (Fig. 1) with several methods (PBE0, LC-wPBE, M062X, and B3LYP) and basis sets (6-31g(,), 6-31++g(,)). All calculations were carried out by the Gaussian 09[15]software. Then we compared the theoretical and experimental values[16]to screen out the optimized method.
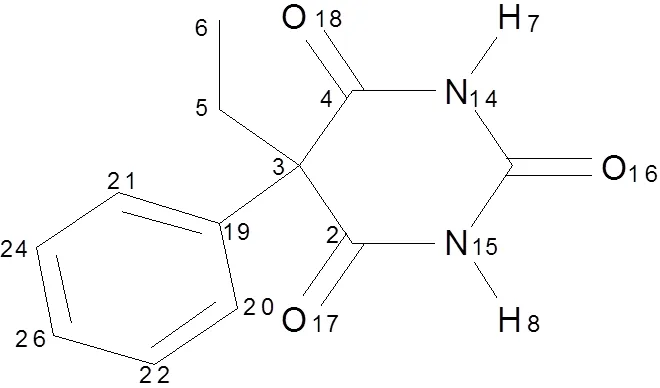
Fig. 1. Chemical structure of PHN
The optimized method was used to calculate the conformations of the template interacting with the monomers. The geometry structures of PHN and VDAT were optimized first of all. Then we calculated the natural bond orbital (NBO)[17]charge and molecular electrostatic potential (MEP) of PHN and VDAT to predict the active sites without imagi- nary frequency. Next, we optimized the compounds of each proportion with the consideration of basis set superposition error (BSSE). In the process of optimi- zation, all isomers of each imprinting proportion were optimized, and we selected the complexes with the lowest binding energy as the research object. Furthermore, the rebinding properties of MIPs toward the template molecule and its analogues were used to study the selective adsorption of MIPs in the molecular imprinting experiment. In the rebinding process, all atomic coordinates of functional mono- mers were fixed in the input file, and then deleted the imprinted molecule. According to different functional groups of the template analogues, the optimized analogues were added in the possible position of cavities. We optimized all the complexes formed by analogues and monomers when the former were located in different positions. The configurations with the lowest energy were selected as the research object. Finally, the AIM theory was employed to reveal the interaction mechanism between PHN and VDAT. The binding energy of PHN with the VDAT was obtained from equation (1):
ΔComplexPHNΣVDAT(1)
In the equation, Δwas the binding energy between PHN and VDAT.Complex,PHN,andVDATwere the single point energies of the complex, PHN and VADT, respectively.
3 RESULTS AND DISCUSSION
3.1 Selection of an appropriate method
The structural parameters of PHN are presented in Table 1. Table 1 shows that deviations between the theoretical and experimental values were all within the margin of error. This result showed that the DFT had some advantages in the aspect of structure optimization. Compared with the M062X and LC- wPBE methods, the deviations of other theoretical methods were larger in bond lengths and angles. For example, the deviations of bond lengths given by PBE0 and B3LYP were 0.002~0.024 and 0.003~0.019 Å, respectively. However, it was in the range of 0~0.015 Å given by the M062X and LC-wPBE methods. Similarly, the deviations of bond angles given by PBE0, B3LYP, and LC-wPBE were larger than that obtained by M062X. In general, the results obtained by the M062X method were closer to the experimental values. In addition, the variations of basis sets can not make significant changes of the molecular structure. As can be seen in Table 1, the differences between the 6-31g(,) and 6-31++g(,) basis sets were 0~0.004 Å and 0~0.7º in bond lengths and angles, respectively. Thus, we chose the M062X/6-31g(,) method to save the computa- tional resources.
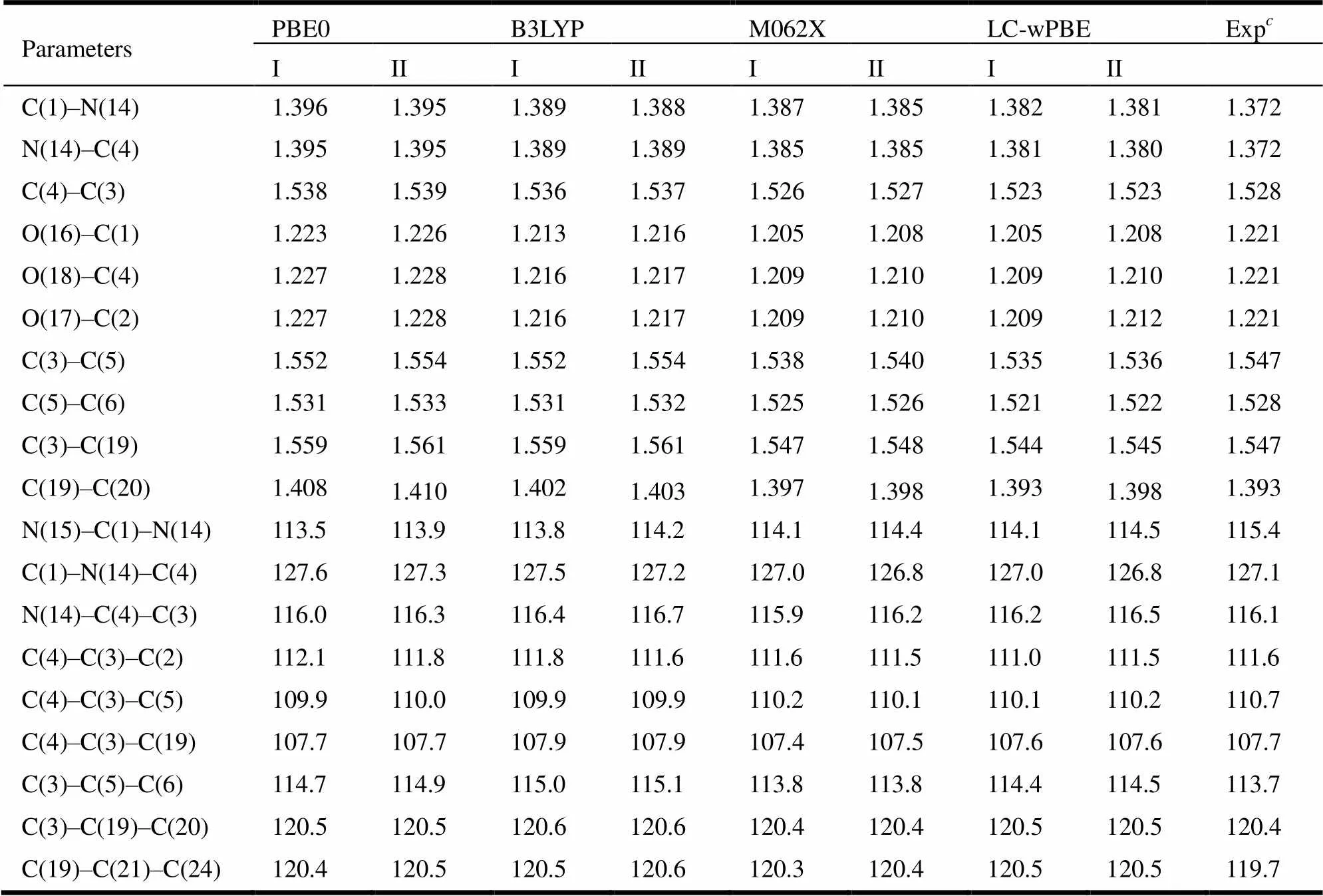
Table 1. Bond Lengths (Å) and Bond Angles (°) Calculated by the PBE0, LC-wPBE, M062X, and B3LYP with Basis Sets (6-31g(d, p), 6-31++g(d, p) and Experimental Values of PHN
6-31g(,),6-31++g(,),Ref. [16]
In order to prove the rationality of the theoretical structure calculated via the M062X/6-31g(,) method further, we simulated the infrared spectrum (Fig. 2) of PHN (Fig. 1). At the same time, we could admire that two N–H stretching vibrations are present around 3109 cm-1in Fig. 2. The peaks around 2974 cm-1belong to the C–H vibrations in benzene ring. The peaks around 1705 cm-1are three strong C=O stretching vibrations. The peaks at 1372 cm-1are the C–H bending vibrations of alkane. The C–H bending vibration peak in benzene can be observed at 697 cm-1and there are many bands around it. All of the theoretical infrared charac- teristic absorption peaks for PHN agreed with the experimental one[18]. Moreover, the M062X method had a good effect on the calculation of molecules[19]. Consequently, we selected the M062X/6-31g(,) method to calculate the properties we need in this paper.
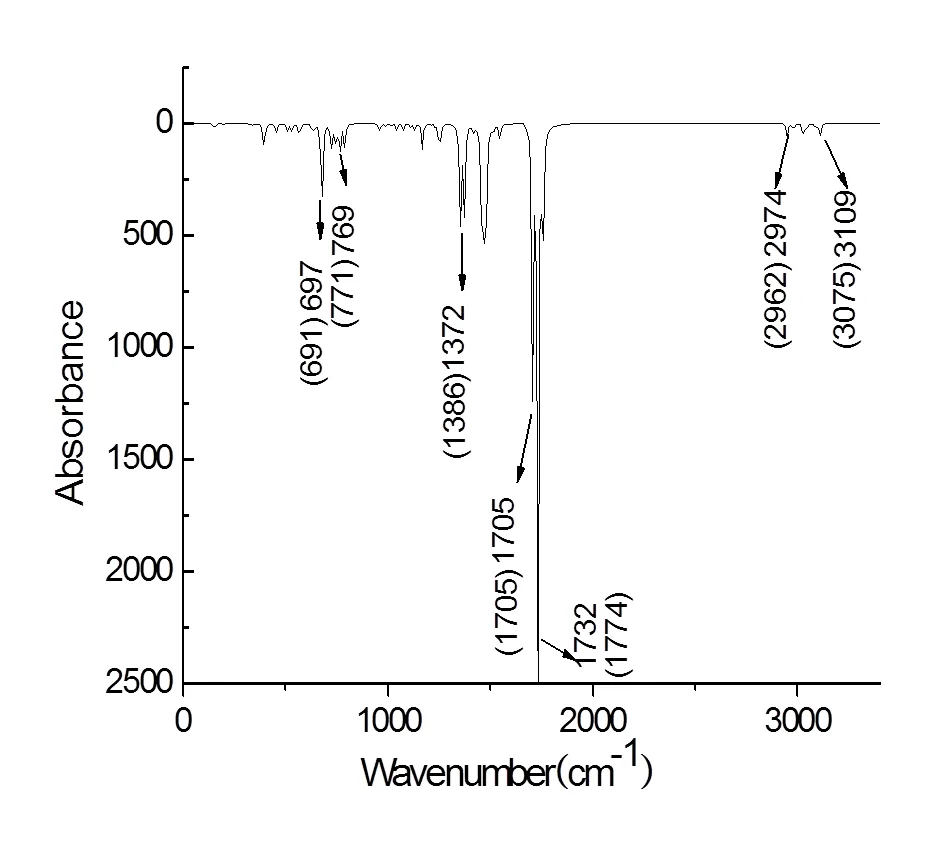
Fig. 2. Simulated infrared spectrum of PHN. The values in brackets were obtained from the experiment
3.2 Optimization and forecast of the active sites for template molecules and functional monomers
In this paper, we studied the atoms’ active sites of molecules on the basis of NBO charge and MEP analysis. From the layout of NBO (Fig. 3a) charge analysis, atoms with more negative charges were the three carbonyl oxygen atoms and two nitrogen atoms of pyrimidine ring, and their negative charge values were O(16):0.600, O(17):0.583, O(18):0.583, N(14):0.720, and N(15):0.720, respectively. Due to the nitrogen atoms belonging to the2hybridiza- tion, it is difficult for these nitrogen atoms to accept a proton. Thus, the three carbonyl oxygen atoms were the main proton acceptors and they were considered as nucleophilic reaction centers. The hy- drogen atoms linked with nitrogen atoms had higher positive charge, with their NBO charges to be H(7): 0.469, and H(8): 0.469, respectively. Therefore, these two hydrogen atoms became the main proton donors. Though the other carbon and hydrogen atoms were not the main proton donors or acceptors, they could not be ignored in the simulation process. Consequently, the active sites of PHN were mainly O(16), O(17), O(18), H(7), and H(8), respectively. The negative charges were mainly distributed on the nitrogen atoms of VDAT (Fig. 3b). Because of the steric factors and arrangement of electropositive and electronegative atoms, the nitrogen atoms of vinyl were easier to approach the PHN than the other nitrogen atoms, so the nitrogen atoms accepting a proton easily of VDAT were N(2):0.611, N(5):0.601 and N(6):0.627. The positive charges were mainly distributed on the hydrogen atoms of sub amino groups, namely, H(8): 0.436, H(9): 0.435, H(11): 0.435, and H(12): 0.437. These atoms became the main proton donors. Thereby, the main active sites of VDAT were H(8), H(9), H(11), H(12), N(2), N(5), and N(6).
The MEP showed the layout of molecular electron cloud. According to the distribution of colors, we could judge the location of strong electropositive and electro negativity atoms so as to determine the active sites. From the scale of MEP in Fig. 3, one can find that the stronger the electronegative of atoms is, the closer to red the color of electron cloud of atoms is, and vice versa, the stronger the electro- positive of atoms is, the closer to blue the color of electron cloud of atoms will be. Obviously, the positive charges of PHN (Fig. 3a) were mainly localized on the hydrogen atoms connected with the two nitrogen atoms of pyrimidine ring. They could easily react with nucleophilic reagent and acquire electrons. The negative charges of PHN were mainly distributed on the carbonyl oxygen atoms in pyrimidine ring. On the contrary with hydrogen atoms, the oxygen atoms couldeasily react with nucleophilic atoms and lose electrons. At the same time, the hydrogen atoms in benzene and ethylene alkyl groups have weak proton-accepting ability. Electronic deficient areas of VDAT were the four hydrogen atoms in the blue region, namely H(8), H(9), H(11), and H(12). The three nitrogen atoms of the three triazole rings were the main proton acceptors.
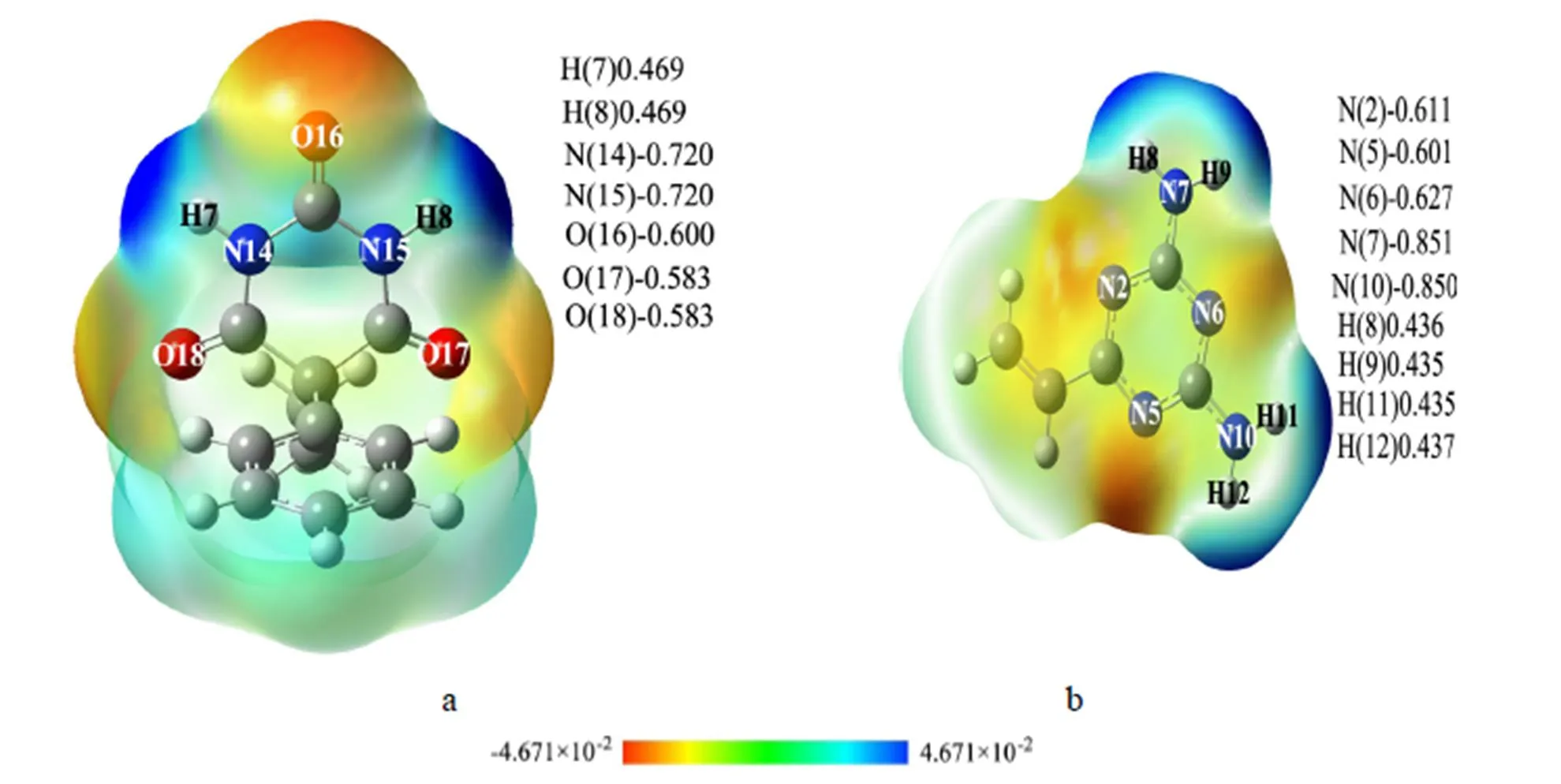
Fig. 3. NBO charges and MEP of PHN and VDAT
3.3 Determination of optimal imprinting ratio between templates and functional monomers
The stabilities of complexes increase as their interaction sites increase. Thus we calculated the interaction sites of complexes, and the results are shown in Table 2, in which the number of hydrogen bonds between PHN and VDAT for ratios (1:1, 1:2, 1:3, 1:4) is 3, 6, 7, and 10, respectively. Thereby, we can see that the interaction between functional monomers and templates became stronger while the number of interactive sites increased, so that the complexes became more stable gradually. When the ratio is increased to 1:5, the number of active sites decreased to 8 and the bond lengths increased slightly. It made the stability of the complexes with this ratio decrease. The interaction strength of template molecule and the functional monomers was a key point for imprinted polymer preparation. Only the linkages of the template molecule and the func- tional monomers were the strongest, could the imprinted molecules be fixed firmly in the process of imprinted polymer preparation and could the functional monomers array accurately in the correct position during the cross link process. Therefore, the appropriate ratio of imprinted molecules was 1:4, which was consistent with those of the binding energies (Δ) for complexes given in Table 2. Namely, the binding energy of the complex with the ratio of 1:4 was the lowest and its stability was the highest. The stable complex configuration is shown in Fig. 4.
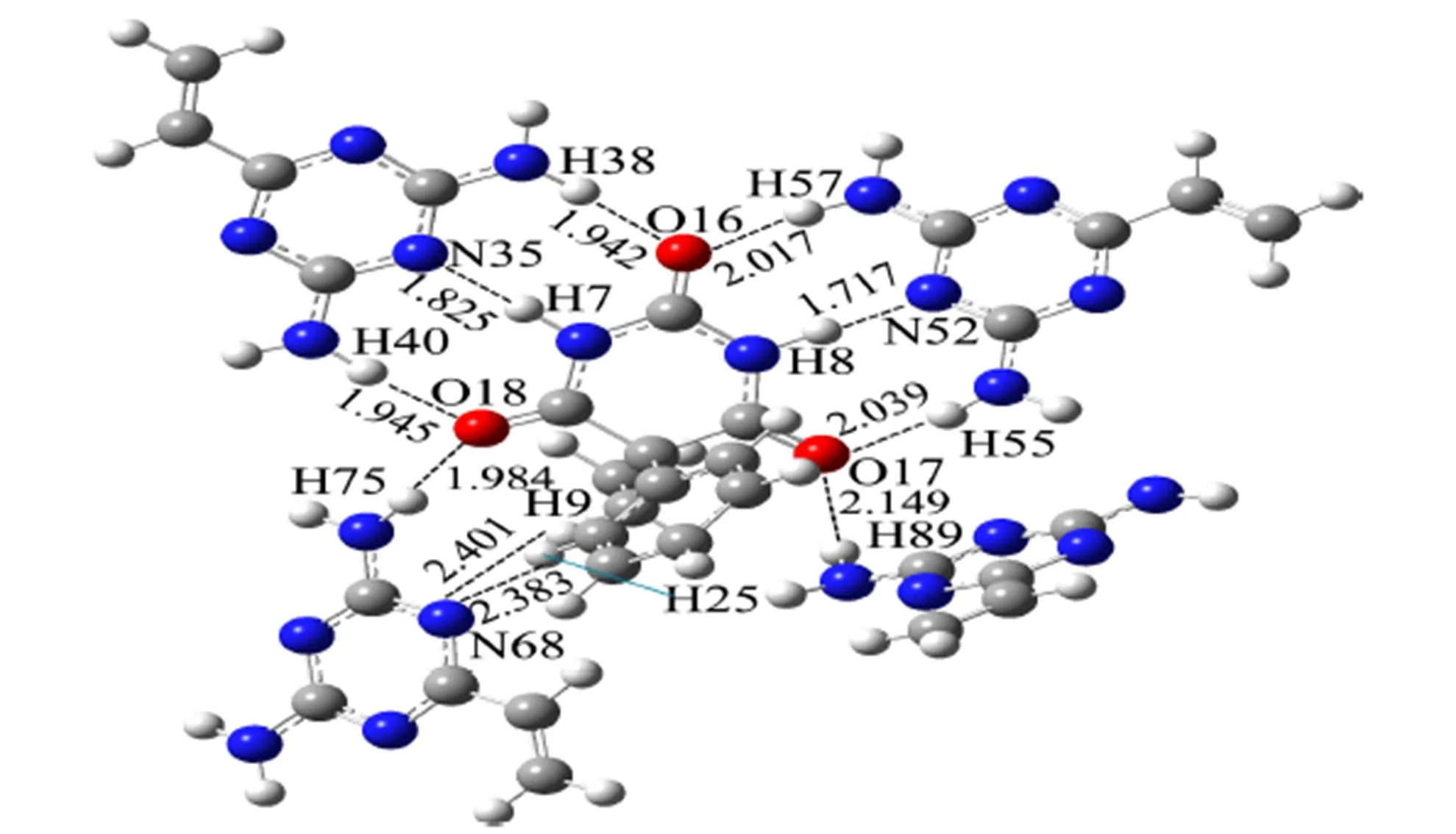
Fig. 4. Model of the complex formed from PHN and VDAT (1:4)
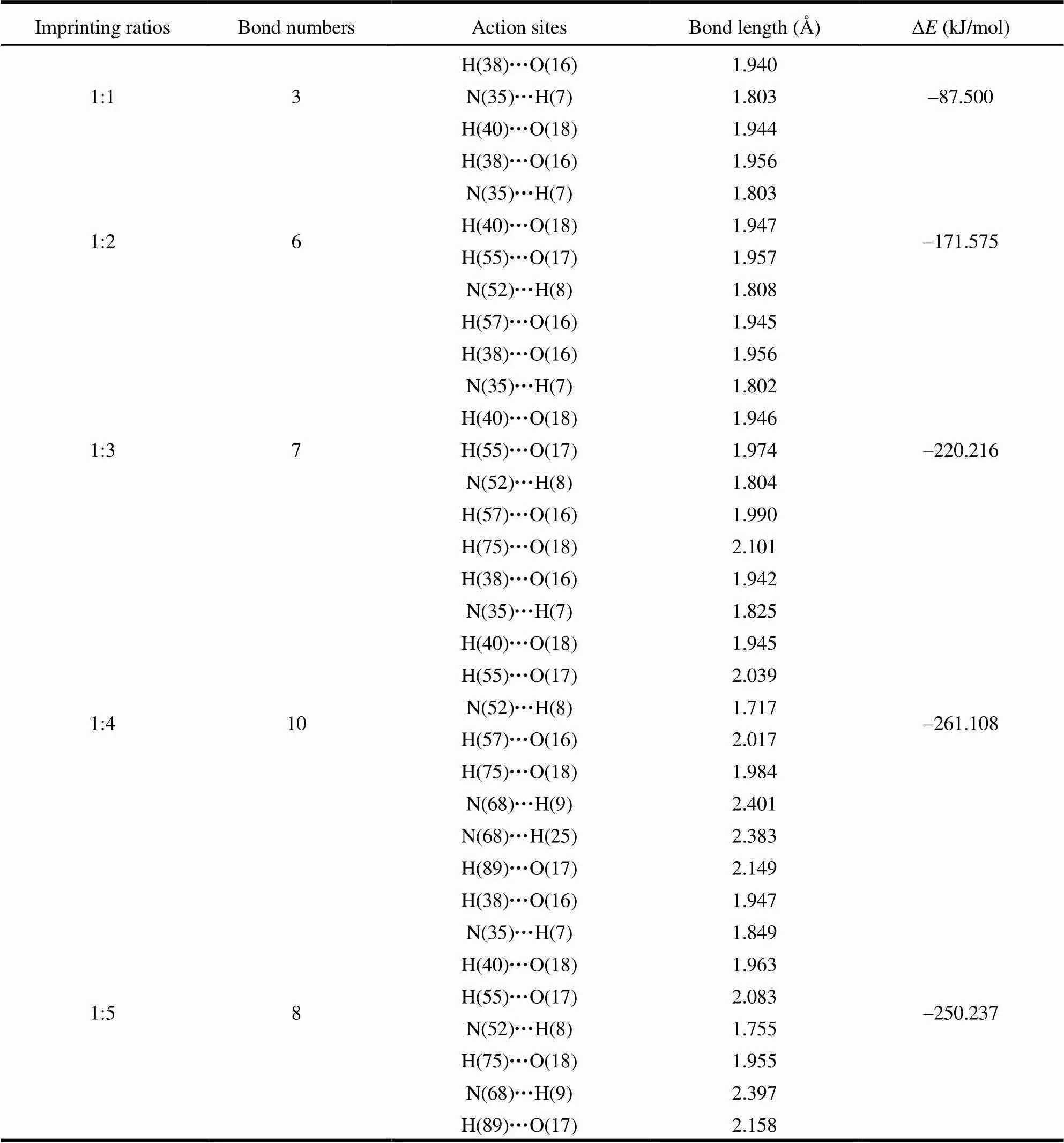
Table 2. Related Parameters of PHN Molecular Imprinting Systems
As could be seen from Fig. 4, carbonyl oxygen atoms as proton acceptors in PHN formed six hydrogen bonds with VDAT, with their active sites and bond lengths as follows: H(38)···O(16) 1.942 Å, H(57)···O(16) 2.017 Å, H(40)···O(18) 1.945 Å, H(75)···O(18) 1.984 Å, H(55)···O(17) 2.039 Å, and H(89)···O(17) 2.149 Å. While the hydrogen atoms as proton donors formed four hydrogen bonds, its active sites and bond lengths were as below: N(35)···H(7) 1.825 Å, N(52)···H(8) 1.717 Å, N(68)···H(9) 2.401 Å, and N(68)···H(25) 2.383 Å. These bond lengths were all in the range of hydrogen bonds[20, 21]. In addition, except the classic hydrogen bonds (N···H and O···H), non-classical hydrogen bonds C6H5···N also existed. The benzene ring was considered as a proton donor and nitrogen atoms as proton acceptors. Double bonds N=C matched the Pi bond in benzene ring. Thus the C–H groups formed strong hydrogen bonds with the lone electron of N atoms.
In order to reveal the polymerization essence of complexes, further, we stimulated the interaction of template and monomers with the help of Multiwfn 3 software[22]based on AIM theory[23]. Bond critical points (BCPs) were used to estimate the interaction between adjacent atoms and these BCPs connected with two atoms respective via two bonds. At the same time, the values of their electron densities ((r)bcp) and Laplacians (▽2(r)bcp) were used to judge the types of interaction force.(r)(r)(r) referred by Rozas[24]was also a useful tool to judge the hydrogen-bonded systems, in which(r) was the Lagrange energy and(r) the Virial po- tential. If ▽2(r)0 and(r) > 0 at BCP, the mole- cules interacted through weak interaction, that was, electrostatic interaction. ▽2(r)0 and(r)0 indicated that the molecules interacted through moderate interactions. ▽2(r)0 and(r)0 demonstrated the presence of strong intermolecular interaction and the interaction force belonged to covalent interaction mostly. The results are shown in Table 3.
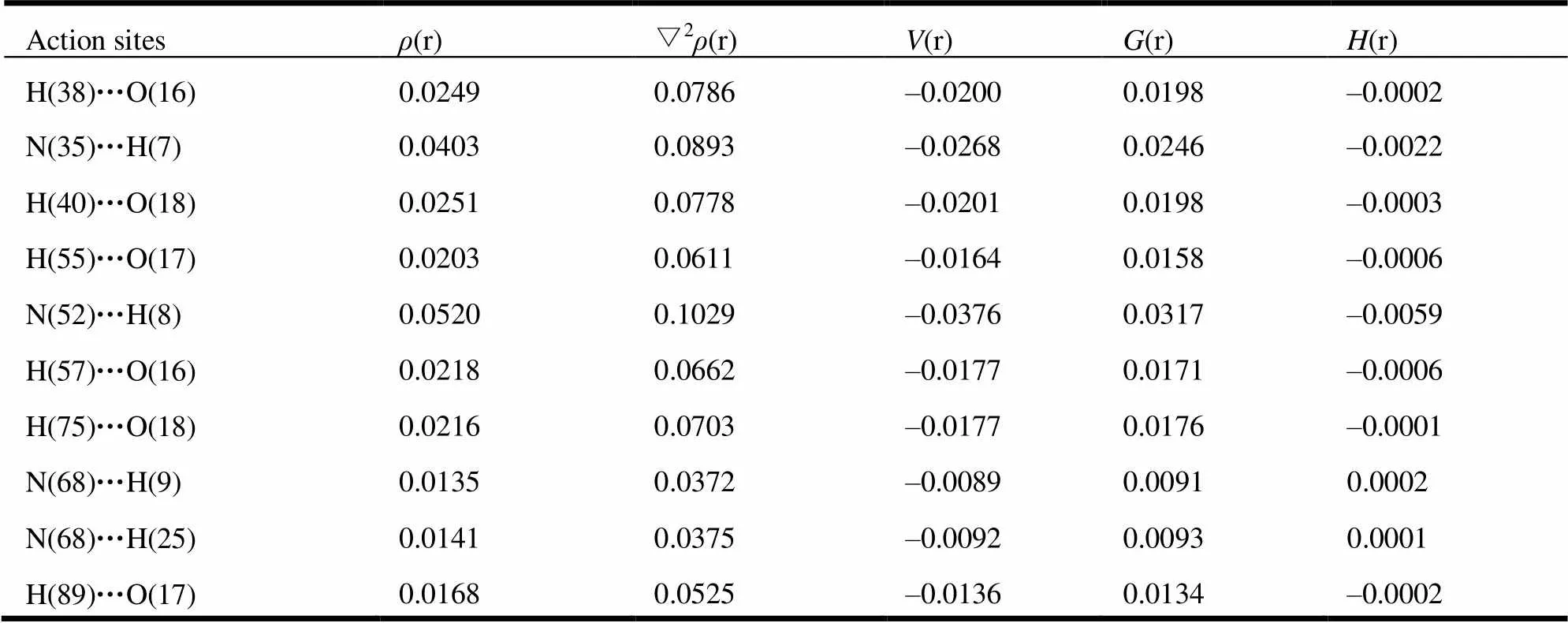
Table 3. AIM Property of the PHN Complex (1:4) (a.u.)
As can be seen in Table 3, the maximum value of(r) was 0.0520 a.u. and the minimum value was 0.0135 a.u. ▽2(r) was in the range of 0.0372~0.1029 a.u. The most hydrogen bonds were all in line with the criteria proposed by Popelier and Bader[25], namely(r) and ▽2(r) were in the ranges of 0.002~0.035 and 0.024~0.139 a.u., respectively. But values of ▽2(r) for N(35)···H(7) and N(52)···H(8) did not fall into the common accepted range referred above. The Laplacians (▽2(r)bcp) values and Virial potential ((r)) values of N(35)···H(7) and N(52)···H(8) were ▽2(r)0 and(r)0. It showed that the interaction of N(35)···H(7) and N(52)···H(8) belonged to medium strength. This result was consistent with that of hydrogen-bond distance mentioned above. The binding distances of N(35)···H(7) and N(52)···H(8) were the shortest (1.825 and 1.717 Å, respectively), and they had the strongest interactions of hydrogen bond.
3.4 Rebinding property research
The rebinding energies of novel imprinted polymers toward the template molecules and its analogues are plotted in Table 4. The results showed that the selective adsorption properties of MIPs were strong obviously and the rebinding energy for PHN was greater than that of analogues, namely the order of rebinding ability was PHNPNTBAR. Compared with the structure of PHN (Fig. 1), PNT (Fig. 5a) and BAR (Fig. 5b) both had the pyrimidine ring and ethyl groups, namely they had some same active sites of hydrogen bond. Thus the MIPs also owned some rebinding properties toward PNT and BAR. The carbon and hydrogen atoms in 3-methyl butyl of PNT and ethyl of BAR had low activity, so their ability of providing protons was weaker than the benzene ring of PHN. Consequently, the rebinding ability of MIPs toward PNT and BAR was weaker than that of PHN. In addition, the difference of molecular structure resulted in the deviations of bond lengths and bond angles between pyrimidine ring and VDAT, so that the imprinted cavity could not match the PNT and BAR perfectly in shape, size, and active sites, and the rebinding energy decreased relatively. Therefore, the recognition sequence of MIPs was PHNPNTBAR after the elution of PHN.
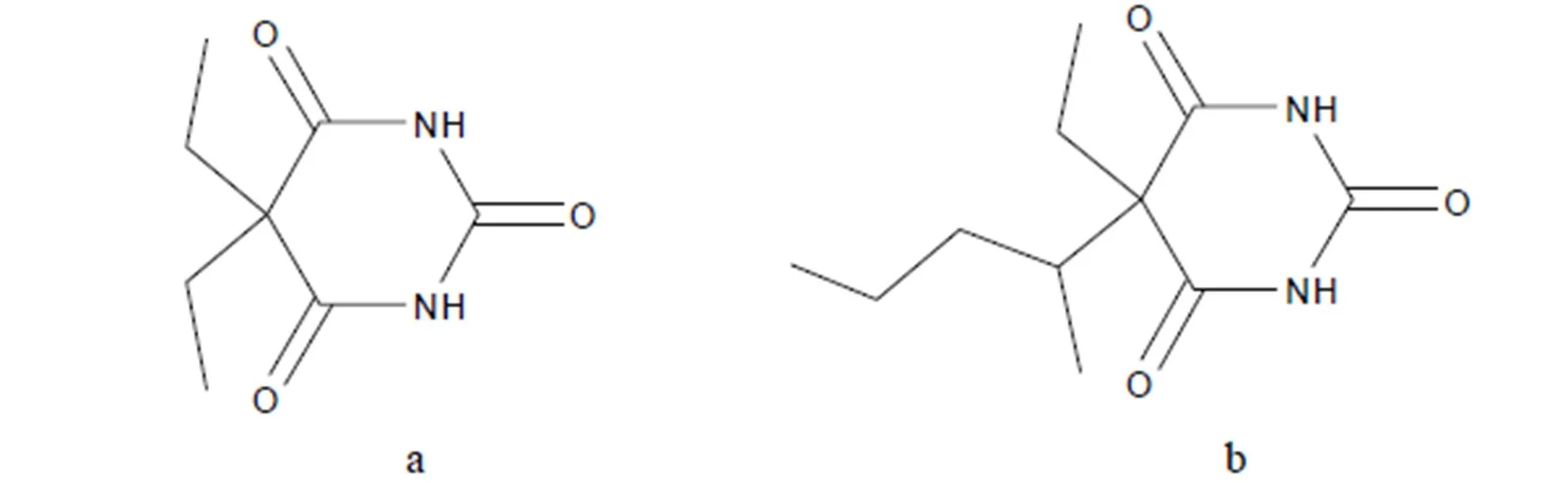
Fig. 5. Molecular structures of the BAR (a) and PNT (b)

Table 4. Rebinding Energies of the Complexes (kJ/mol)
4 CONCLUSION
In this work, PHN was taken as a template mole- cule and VDAT as a functional monomer. The hybrid density functional method (M062X) was used to discuss hydrogen bonding situation of PHN-MIPs, and the AIM was applied to reveal the nature of bond between PHN with VDAT. The results showed that PHN and VDAT interacted through hydrogen bonds. There were 10 hydrogen bonds in the cavity of stable complexes when the ratio was 1:4. Recognition sequence of MIPs toward PHN, PNT, and BAR was PHNPNTBAR after the elution of PHN. This work can help to choose the imprinting ratio, and the selective study also provided a theore- tical basis for the synthesis of universal molecularly imprinted materials. Furthermore, it can also supply systemic theoretical reference to improve the molecular imprinting technique.
(1) Lian, Z. R.; Wang, J. T. Molecularly imprinted polymer for selective extraction of malachite green from seawater and seafood coupled with high-performance liquid chromatographic determination.2012, 64, 2656-2662.
(2) Liang, R. N.; Kou, L. J.; Chen, Z. P.; Qin, W. Molecularly imprinted nanoparticles based potentiometric sensor with a nanomolar detection limit.. 2013, 188, 972-977.
(3) Nestić, M.; Babić, S.; Pavlović, D. M.; Sutlović, D. Molecularly imprinted solid phase extraction for simultaneous determination of Δ9-tetrahydrocannabinol and its main metabolites by gas chromatography–mass spectrometry in urine samples.2013, 231, 317-324.
(4) Zhang, Y. Q.; Shan, X.; Gao, X. Q. Development of a molecularly imprinted membrane for selective separation of flavonoids.. 2011, 76, 337-344.
(5) Florian, M.; Branka, S.; Denise, R.; Boris, M. Computational and experimental study on the influence of the porogen on the selectivity of 4-nitrophenol molecularly imprinted polymers.2012, 744, 68-74.
(6) Wyszomirski, M.; Prus, W. Molecular modelling of a template substitute and monomers used in molecular imprinting for aflatoxin B1 micro-HPLC analysis.. 2012, 38, 892-895.
(7) Sun, J. N.; Liu, J. B.; Tang, S. S.; Jin, R. F. Theoretical researches on the self-assembly system of ciprofloxacin imprinted polymers.. 2013, 32, 1204-1210.
(8) Djozan, D.; Ebrahimi, B. Preparation of new solid phase micro extraction fiber on the basis of atrazine-molecular imprinted polymer: application for GC and GC/MS screening of triazine herbicides in water, rice and onion.2008, 616, 152-159.
(9) Liang, R. N.; Zhang, R. M.; Qin, W. Potentiometric sensor based on molecularly imprinted polymer for determination of melamine in milk.2009, 141, 544-550.
(10) Liu, J. B.; Sun, J. N.; Tang, S. S.; Chen, K. Y.; Jin, R. F. Theoretical researches on the recognizing characteristics of atrazine imprinted polymers with different functional monomers.. 2012, 31, 1794-1802.
(11) Goar, S. S.; Cristina, T.; Ibon, A.; José, E. Electron density shift description of non-bonding intramolecular interactions.2012, 991, 124-133.
(12) Masahiko, H.; Katsuhiko, H. Pair density functional theory.. 2013, 1003, 91-96.
(13) Lu, L. L.; Hu, H.; Hou, H.; Wang, B. S. An improved B3LYP method in the calculation of organic thermochemistry and reactivity.2013, 1015, 64-71.
(14) Zhang, J. H.; Wang, X. J. Developments on correction model of density functional theory based on statistics methods.1996, 25, 223-230.
(15) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery, J. A.; Stratmann, R. E.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Baroe, V.; Cossi, M.; Cammi, R.;Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski, J.; Ortiz, J. V.; Baboul, A. G.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.;Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P. M. W.;Johnson, B.; Chen, W.; Wong, M. W.; Andres, J. L.; Gonzalez, C.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A.Pittsburgh PA: Gaussian Inc. 2009.
(16) Platteau, C.; Lefebvre, J.; Hemon, S.; Baehtz, C.; Danede, F. Structure determination of forms I and II of phenobarbital from X-ray powder diffraction.2005, 61, 80-88.
(17) Carpenter, J. E.; Weinhold, F. Analysis of the geometry of the hydroxymethyl radical by the “different hybrids for different spins” natural bond orbital procedure.1988, 169, 41-62.
(18) Cai, X. L.; Wu, G. P. Spectra research on the identification of mixture medicine of barbitals.2001, 18, 563-567.
(19) Zhang, Y.; Xu, X. A new generation density functional XYG3.2012, 24, 1023-1037.
(20) Ranganathan, A.; Kulkarni, G. U.; Rao, C. N. R. Probing the hydrogen bond through experimental charge densities.2003, 656, 249-263.
(21) Siodłak, D.; Bujak, M.; Staś, M. Intra- and intermolecular forces dependent main chain conformations of esters of,-dehydroamino acids.2013, 1047, 229-236.
(22) Lu, T. Multiwfn: multifunctional wave function analyzer, Version 2.31. 2012.
(23) Bader, R. F. W. A bond path: a universal indicator of bonded interactions.1998, 102, 7314-7323.
(24) Roas, I.; Alkorta, I.; Elguero, J. Behavior of ylides containing N, O, and C atoms as hydrogen bond acceptors.2000, 122, 11154-11161.
(25) Popelier, P. L. A.; Bader, R. F. W. The existence of an intramolecular C–H…O hydrogen bond in creatine and carbamoyl sarcosine.1992, 189, 542-548.
28 April 2014;
6 June 2014
the Natural Science Foundationof Jilin Province (No.201215180), the Science and Technology Developmental Plan of Jilin Province(No.20130206099SF), the Science and Technology Research Projects for EducationDepartment of Jilin Province (No.201359), and the National Natural Science Foundation of China (No. 21302062)
. Liu Jun-Bo, born in 1964, professor. Tel: 18604309293. E-mail: liujb@mail.ccut.edu.cn Tang Shan-Shan. Born in 1981, doctor. Tel: 13604360743. E-mail: tangshanshan81@163.com

- 结构化学的其它文章
- Thermoelectric Properties of the CuGaTe2 Crystal from First-principles Calculations: the Role of Doping and Temperature①
- Morphology, Size-controlled Synthesis of CoO anostructure and Its Magnetic Property①
- The First Hybrid Wells-Dawson-type Polytungstate Monosupported by Cd-coordination Complex via Di-bridging O-atom①
- Synthesis, Crystal Structure and Antiproliferative Activity of 2-(((5-(((5,7-Dimethyl-[1,2,4]triazolo-[1,5-a]pyrimidin-2-yl)thio)methyl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)methyl)-4H- chromen-4-one Methanol Solvate①
- Synthesis, Crystal Structure and Luminescent Property of an Eu3+ Complex①
- Synthesis, Dimer Crystal Structure and Herbicidal Activity of 2-(4-Ethoxybenzoyl)cyclopentane-1,3-dione①