
抗凝血酶基因10381T缺失导致的Ⅰ型抗凝血酶缺陷症
2014-11-27 07:27李正民宫璀璀吕金利方盼盼王广兰伦永志
基础医学与临床 2014年3期
李正民,宫璀璀,吕金利,方盼盼,王广兰,伦永志,白 洁
(1.郑州大学 第三附属医院 科研中心, 河南 郑州 450000;2.中国人民解放军第153中心医院 检验科, 河南 郑州 450042;3.济南军区普外科中心, 河南 郑州 450000; 4.新乡医学院 研究生院,河南 新乡 453000;5.大连大学 医学院医学研究中心,辽宁 大连 116000;6.中国人民解放军总医院 北京301医院 全军检验中心, 北京 100000)
抗凝血酶基因10381T缺失导致的Ⅰ型抗凝血酶缺陷症
李正民1,2,宫璀璀1,2*,吕金利3,方盼盼1,王广兰2,4,伦永志5,白 洁6
(1.郑州大学 第三附属医院 科研中心, 河南 郑州 450000;2.中国人民解放军第153中心医院 检验科, 河南 郑州 450042;3.济南军区普外科中心, 河南 郑州 450000; 4.新乡医学院 研究生院,河南 新乡 453000;5.大连大学 医学院医学研究中心,辽宁 大连 116000;6.中国人民解放军总医院 北京301医院 全军检验中心, 北京 100000)
目的对1例遗传性抗凝血酶(AT)缺陷症先症者及其家系进行表型诊断和基因诊断,并探讨其家系成员发病机制。方法用发色底物法检测该家系9名成员的AT活性(AT∶A)、蛋白S活性(PS∶A)、蛋白C活性(PC∶A),用免疫比浊法检测AT抗原量(AT∶Ag),用Western blot检测血浆中的AT分子质量和含量,抽提外周血基因组DNA,用PCR对AT基因的7个外显子及其侧翼序列进行扩增,用直接测序法对该家系所有成员的扩增产物进行测序分析并进行基因突变检测,同时筛查100例正常人以排除基因突变的多态性。结果该家系先症者AT∶A 和AT∶Ag分别为48%和121 mg/L,先症者AT基因的第6外显子发现10381T del。其家系的部分成员检测到相同的移码突变。结论该家系先症者及部分成员存在Ⅰ型遗传性抗凝血酶缺陷症,是由AT基因10381T del移码突变所致。
抗凝血酶;基因突变;易栓症
人体的抗凝系统主要有抗凝血酶(antithrombin, AT)系统(包括抗凝血酶和肝素等)和蛋白C系统(包括蛋白C, 蛋白S等),抗凝系统的功能异常可导致血栓形成倾向增高,引起易栓症。常见的遗传性易栓症有蛋白S缺陷症、蛋白C缺陷症、抗凝血酶缺陷症、抗活化的蛋白C症(FV Leiden突变)、凝血酶原20210A突变等。抗凝血酶(AT)是一种丝氨酸蛋白酶(serpins)抑制剂,主要具有抑制丝氨酸蛋白酶活性的凝血酶以及Ⅶa、Ⅸa、Ⅹa、Ⅺa、Ⅻa等因子,是人体最重要的抗凝因子之一,约占抗凝活性的70%左右,在人体抗凝系统中发挥着重要作用。遗传性抗凝血酶缺陷症是遗传性易栓症的一种,是由于编码抗凝血酶的基因发生突变所引起的一种常染色体显性遗传病,在人群中的发病率为万分之二到万分之五[1-2]。现对一例抗凝血酶缺陷症家系进行了家系调查、临床表型诊断、实验诊断和基因诊断,发现了一个10381T del移码突变。
1 资料与方法
1.1 家系资料
先症者,男,汉族,51岁,1981年首次出现下肢静脉血栓,后反复多次出现下肢静脉血栓,2011年5月因腹痛入院,经剖腹探查发现肠系膜静脉血栓,行外科手术治疗,2012年3月因右下肢肿胀疼痛再次入院治疗,经血管造影诊断为右下肢深静脉血栓形成,后口服华法林治疗半年,病情好转。现定期监测凝血机制,以预防性治疗为主。家族史阳性,家系3代10人中,其母亲、两个姐姐、侄子均有下肢静脉血栓史,家系其他成员无类似病史。家系图见图1。

图1 遗传性AT缺陷症家系图Fig 1 Hereditary AT deficiency pedigree chart
1.2 标本采集与相关指标检测
在获得该家系所有成员的知情同意后,对家系成员9人分别采用负压静脉采血,枸橼酸钠抗凝管采血2.7 mL(1∶9抗凝)2管、干燥管采血5 mL、乙二胺四乙酸(EDTA)管采血2 mL。立即4 ℃ 3 000 r/min离心10 min(除EDTA管),收集1管部分枸橼酸钠抗凝血浆冻存-80 ℃,其他样本分别进行血液细胞分析、凝血4项(PT、APTT、TT和FIB;)、D-D二聚体检测(Stago公司)、肝功能、肾功能、同型半胱氨酸(HCY)含量检测(四川迈克生物科技股份有限公司)、狼疮抗凝物(LA)、抗心磷脂抗体检测(北京贝尔公司)、蛋白C活性(PC:A)、蛋白S活性(PS:A)、AT活性检测(采用发色底物法,Dade Behring公司)、AT抗原量检测(采用免疫比浊法, Beckman Coulter公司)等。
1.3 血浆中AT分子质量和含量检测
采用Western blot检测:将该家系成员血浆20倍稀释(用磷酸盐缓冲液-PBS),与上样缓冲液3∶1混合,放入100 ℃沸水中加热5 min。用10% SDS-PAGE电泳(60 V,55 mA,3.5 h)后,经湿转法(60 V,160 mA,3 h)将蛋白条带转移至硝酸纤维素膜上,用5%脱脂奶粉(脱脂奶粉0.5 g,TBST 10 mL)封闭1 h, 用一抗(antithrombin Ⅲ rabbit monoclonal antibody, EPIT MICS,An Abcam公司)孵育2 h,彻底清洗后用二抗(HRP标记的goat anti-rabbit IgG,上海生工公司分装)孵育。彻底清洗后用DAB显色液(北京中杉金桥公司)显色10 min。蛋白质分子量标记采用彩色预染蛋白质Marker(10~180 kd)。
1.4 基因组DNA抽提
用该家系成员外周血(枸橼酸钠抗凝),采用DNA提取试剂盒(Fermentas公司)提取全基因组DNA,严格按照试剂说明书操作提取。
1.5 PCR扩增及产物纯化
使用Primer5引物设计软件设计AT基因第3外显子及其侧翼序列的扩增引物,第1、2、4、5、6和7外显子的扩增引物序列参照文献报道[3],引物信息见表1。PCR总体系为25 μL:1×缓冲液,dNTP终浓度为0.25 mmol/L,MgCl2终浓度为1.5 mmol/L,引物终浓度为0.2 μmol/L,模板DNA 50 ng,Taq酶为1.25 U(TIANGEN公司)。反应条件为:95 ℃预变性5 min,然后95 ℃ 30 s,57 ℃退火30 s(第3外显子采用62 ℃退火,第5外显子采用60 ℃退火),72 ℃延伸30s,30个循环,72 ℃延伸10 min。PCR产物80 μL,加20 μL电泳缓冲液(4∶1),混匀后进行琼脂糖电泳(18 g/L琼脂糖,含溴化乙锭),在254 nm紫外灯下切取相应条带,用QIAquick Extracion Kit试剂盒进行PCR产物回收、纯化(QIAGEN公司)。
1.6 AT基因测序及突变分析
PCR纯化产物由上海生工生物工程有限公司进行正向和反向测序,测序仪器为ABI公司3730xl基因分析仪,试剂为BigDye terminator v3.1,测序图谱分析采用Chromas软件。用BLAST在线工具将该家系成员AT基因测序结果与AT基因序列(Genbank:X68793.1)进行比对,分析基因突变。将AT基因第6外显子(存在10381Tdel)经再次PCR扩增、纯化、双向测序,再次进行基因突变验证分析。
1.7排除基因多态性及FVLeiden突变和凝血酶原G20210A突变
针对该家系突变位点,对100名正常人进行筛查:抽取枸橼酸钠抗凝血2 mL,提取DNA,PCR扩增相应突变位点,测序后进行相应位点突变分析,以排除基因多态性。对先症者FV Leiden突变检测:引物为F, 5′-CCATTATTTAGCCAGGAGACC 3′,R, 5′-ATTGGTTCCAGCGAAAGC 3′;对外显子10及侧翼序列进行扩增(退火温度60 ℃)、测序。对先症者凝血酶原G20210A突变检测:引物为F,5′-TCTAG AAACAGTTGCCTGGC 3′, R, 5′-TCCAGTAGTATT ACTGGCTC 3′;对外显子14及侧翼序列进行扩增(退火温度53 ℃)、测序。
2 结果
2.1 易栓症初筛检测结果
先症者检测结果 AT:A 48.4%,AT:Ag 121 mg/L,PS:A 164%,PC:A 100.4%,PT 14.6s,INR 1.21,APTT 35.8 s,TT 14.0 s,FIB 3.38 g/L。该家系9人中有5人(先症者及其母亲、两个姐姐、侄子)表现为AT∶Ag和AT∶A的降低,而蛋白C和蛋白S活性正常。该家系成员的血液细胞分析、凝血4项(PT、APTT、TT和FIB)、肝功能和肾功能无明显异常,D-D二聚体、抗心磷脂抗体、LA和同型半胱氨酸检测结果均正常。
2.2 血浆AT蛋白Western blot检测结果
该家系成员的AT蛋白在58 ku区域处存在与正常对照分子量一致的特异性条带,但该家系中有5人(含先症者)的AT蛋白条带阳性强度明显减弱(分别为Ⅱ1、Ⅱ2、Ⅱ3、Ⅰ2和Ⅲ1)(图2)。

表1 AT基因PCR引物序列及扩增区域Table 1 AT gene PCR primers and amplified region

图2 该家系AT蛋白Western blot检测结果Fig 2 The pedigree AT protein Western blot test results
2.3 基因检测分析
先症者的AT基因第6外显子存在一处缺失,即:10381 Tdel,引起了移码突变,导致该位点之后的编码序列发生改变,造成翻译的第400位到403位4个氨基酸被不同氨基酸替代,分别为Phe400Ser,His401Ile,Lys402Arg,Ala403his,第405位亮氨酸之后提前终止编码,第406位到最后464位氨基酸出现缺失;该家系中还有4名成员的AT基因第6外显子存在10381 Tdel,与先症者的AT基因存在相同突变,测序图见(图3)。
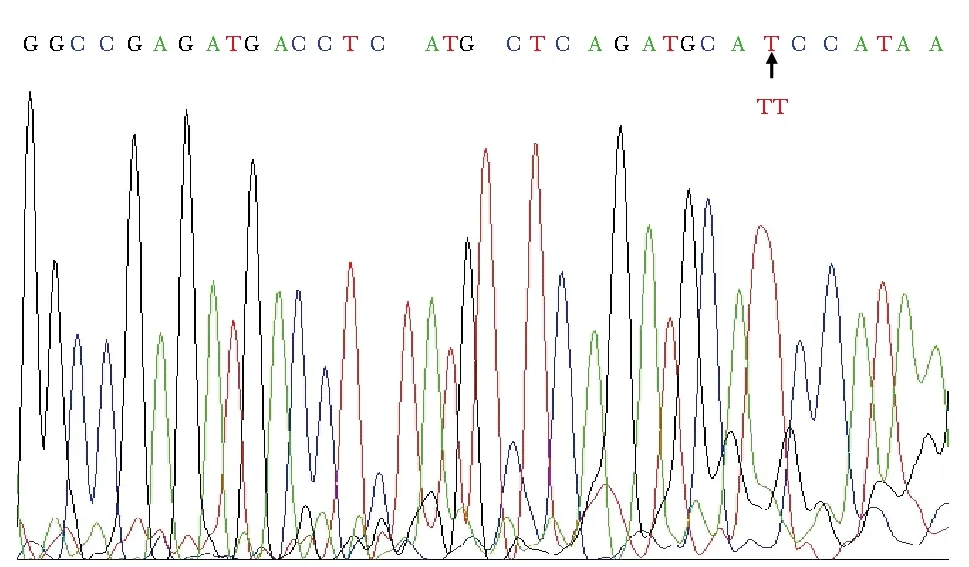
图3 先症者第6外显子部分测序结果Fig 3 The proband’s exon sixth part of sequencing results(positive sequence, arrow indicates the deletion site)
2.4AT基因突变位点多态性排除及FVLeiden突变和凝血酶原G20210A突变检测
对于该家系发现的AT基因突变位点,通过对100名正常人的相应位点测序分析,未发现相同突变,可以排除该突变位点的基因多态性。对先症者的FV基因第10外显子和凝血酶原基因第14外显子进行PCR扩增、产物测序,未发现FV Leiden突变和凝血酶原G20210A突变。
经基因数据库(http://www1.imperial.ac.uk/departmentofmedicine/divisions/experimentalmedicine/haematology/coag/antithrombin/type1/.)检索后发现,该AT基因10381 Tdel杂合突变在国内外未发现有相同位点突变的报道。国内报道的AT基因缺失突变较少,已报道了3例,本例为第4例。
3 讨论
AT能够灭活凝血酶、FXa等因子,具有抗凝血作用,与肝素结合后可使抗凝血作用增加数千倍以上[4]。有研究报道[5]: AT有两个重要的功能区,一个是位于羧基(C)端的反应位点,能够灭活凝血酶; 另一个是位于氨基(N)端的肝素结合区。有研究显示,AT在血液中的浓度较低,此时的抗凝活性也低,当有肝素分子存在的情况下,能够与肝素中的戊糖(pentasaccharide)序列结合,AT的构象发生改变,反应中心环(RCL)暴露,能够最有效的发挥抗凝作用[6-7]。
AT基因的突变可以导致AT蛋白的分泌障碍、胞内滞留而不能发挥抗凝作用,还可以使AT的反应位点异常变异而致活性降低,或者使AT的肝素结合位点异常变异而致与肝素亲和力降低。AT缺陷症临床表现为反复形成静脉血栓,如肠系膜静脉、髂静脉和下肢深静脉等。遗传性AT缺陷症在临床上可分为两型:Ⅰ型,是由于AT合成障碍,表现为AT含量的减少和AT活性的降低;Ⅱ型,是由于AT结构的异常,仅表现为AT活性的降低。缺失突变是一种由于基因组DNA多核苷酸链中碱基对的缺失,以致自缺失点之后部分或所有的三联体遗传密码子组合发生改变的基因突变形式。据报道:法国的两个家族中AT的外显子3A上分别出现小移码缺失,因此引起患者的AT基因缺乏翻译,AT分泌减少,导致两家族多成员发生血栓栓塞症[8]。关于多部位的缺失,有报道发现了AT基因组12个部位的缺失,这些缺失突变造成终止密码子的形成,转录提前停止,从而影响AT肝素结合位点和反应位点的形成,降低了AT活性[9]。
该家系先症者反复多次出现下肢静脉血栓,近年来出现肠系膜静脉血栓,经基因分析的诊断结果显示存在AT基因10381T缺失,且排除获得性易栓症、FV Leiden突变和凝血酶原G20210A突变。该缺失突变导致AT读码框架移位,第399位丙氨酸之后编码表达了4个不同的氨基酸(S、I、R和H替代了F、H、K和A),导致氨基酸序列的改变,且第405位亮氨酸之后提前出现终止密码子,蛋白质表达提前终止,仅编码了405个氨基酸(正常AT蛋白由464个氨基酸组成),用Spdbv软件分析发现,空间位阻增加,AT构象发生改变,影响了反应中心环的暴露。先症者的母亲及两个姐姐、侄子都存在相同位点的缺失突变,均有不同程度的静脉血栓形成史,根据以上相应检查结果可明确诊断该家系存在遗传性AT缺陷症Ⅰ型。
综上所述,AT基因10381 Tdel杂合突变在国内外未发现相同位点缺失的报道,该研究对该家系易栓症的明确诊断、治疗提供了基因诊断依据和理论支持,为以后的疾病预防干预、减少血栓性疾病所带来的身心痛苦和财产损失提供帮助,为遗传性易栓症的分子诊断、发病机制研究提供资料。
[1] Fischer R, Sachs UJ, Heidinger KS,etal. Prevalence of hereditary antithrombin mutations is higher than estimated in patients with thrombotic events[J].Blood Coagul Fibrinolysis, 2013,24:444-448.
[2] Cooper PC, Coath F, Daly ME,etal. The phenotypic and genetic assessment of antithrombin deficiency[J].Clin chem lab med, 2010,48: 67-78.
[3] 傅启华,许先国,丁秋兰,等. 抗凝血酶基因13389G缺失导致的Ⅰ型抗凝血酶缺乏症[J].中华血液学杂志,2002,23:588-590.
[4] Izaguirre G,Swanson R,Raja SM,etal.Mechanism by which exosites promote the inhibition of blood coagulation proteases by heparin activated antithrombin[J]. J Biol Chem, 2007,282:33609-33622.
[5] Patna Ikmm, Moll S. Inherited antithrombin deficiency: a reiew [J].Haemophilia,2008,14:1229-1239.
[6] Johnson DJ, Liw, Adams Te,etal. Antithrombin S195A factor Xaheparin structure reveals the allosteric mechanism of antitrombin activation[J].EMBO J, 2006,25:2029-2037.
[7] Muszbek L, Bereczky Z, Kovacs B,etal. Antithrombin deficienfy and its laboratory diagnosis[J].Pubmed-indexed for MEDLINE,2013,PMID:2106-2218.
[8] Veronique Picard, Alessundra Bura, Josep Emmericn,etal. Molecular bases of antithrombin deficiency in French families:identification of seven novel mutations in the antithrombin gene[J]. Bri J Haematol, 2000,110:731-734.
[9] J.Corral, J.A.Huntington, R.Gonzalez-conejero,etal. Mutations in the shutter region of antithrombin result in formation of disulfide-linked dimers and severe venous thrombosis[J].J Thrombo Haemost,2004,2:931-939.
Type Ⅰ antithrombin deficiency due to 10381T deletion in antithrombin gene
LI Zheng-min1,2, GONG Cui-cui1,2*, LÜ Jin-li3, FANG Pan-pan1, WANG Guang-lan2,4,LUN Yong-zhi5, BAI Jie6
(1.Research Center, the third Affiliated Hospital, Zhengzhou University, Zhengzhou 450000; PLA No.153 Hospital;2.Ji’nan Military Region Inspection Center; 3.General Surgery Center, Ji’nan Military Region, Zhengzhou 450000;4.Graduate School,Xinxiang Medical College, Xinxiang 453000; 5.Medical Research Center, Dalian University School of Medicine,Dalian 116000;6.PLA Inspection Center,Beijing 301 Hospital-PLA General Hospital, Beijing 100000,China)
ObjectiveTo make a phenotype diagnosis and gene diagnosis aiming directly at one case of hereditary antithrombin (AT) deficiency syndrome of proband and their family phenotype, and explore the pathogenesis of family members.MethodsThe activity of AT(AT∶A), protein S and protein C were detected by chromogenic substrate method, with immune turbidimetry on AT antigen (AT∶Ag) detection, the molecules weight and content of AT were detected by Western bloting method, Genomic DNA was extracted from blood, the 7 exons of AT and flanking sequences were amplified by PCR, products of PCR of all family members were conducted by direct sequencing analysising and gene mutation detection, screening 100 cases of normal people to exclude the of polymorphism of gene mutation.ResultsThe AT∶A and AT∶Ag of proband was 48% and 121 mg/L respectively, the proband’sixth exon of AT gene is c.10381T del. Some members of the family were detected the same frameshift mutations.
ConclusionsThe pedigree and some members of the first symptoms are type Ⅰ hereditary antithrombin deficiency due to AT gene 10381T del frameshift mutations.
antithrombin; gene mutation; thrombophilia
2013-09-02
2013-11-27
*通信作者(correspondingauthor): gongcuijin@yahoo.com.cn
1001-6325(2014)03-0397-05
研究论文
R 394.3
A
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
上海金属(2021年6期)2021-12-02
昆明医科大学学报(2021年3期)2021-07-22
中国生殖健康(2020年4期)2021-01-18
中华养生保健(2020年7期)2020-11-16
生物学通报(2019年3期)2019-02-17
中国生殖健康(2018年4期)2018-11-06
现代检验医学杂志(2016年2期)2016-11-14
中国生化药物杂志(2015年4期)2015-07-07
湖北农业科学(2014年11期)2014-09-10
