
无雄激素条件下表皮生长因子诱导前列腺癌细胞系增殖及雄激素受体磷酸化
2014-11-27 07:27孙雪飞白雪燕周永建刘元波
基础医学与临床 2014年3期
孙雪飞,赵 晖,李 扬,钱 筠,白雪燕,周永建,朱 红,张 勇,刘元波,2*
(首都医科大学 附属北京天坛医院 1.血液肿瘤科;2.输血科;3.泌尿外科,北京,100050)
无雄激素条件下表皮生长因子诱导前列腺癌细胞系增殖及雄激素受体磷酸化
孙雪飞1#,赵 晖2#,李 扬1,钱 筠1,白雪燕1,周永建3,朱 红1,张 勇3,刘元波1,2*
(首都医科大学 附属北京天坛医院 1.血液肿瘤科;2.输血科;3.泌尿外科,北京,100050)
目的探讨在无雄激素的条件下,表皮生长因子对前列腺癌细胞增殖及雄激素受体磷酸化的影响。方法以LNCaP 及LAPC4 AR为研究对象,在无雄激素的条件下,EGF处理后, Western blot方法测定LNCaP 及LAPC4 AR磷酸化状态;siRNA转染方法敲除Src基因或Ack1基因,观察对AR磷酸化的影响;CCK-8检测细胞增殖;定时定量RT-PCR检测前列腺特异抗原及人体激肽释放酶2 mRNA表达。结果EGF通过Src激酶和另一未知激酶导致AR Tyr-534及AR Tyr-267特异位点磷酸化;相比未用EGF对照组,EGF能够促进前列腺癌细胞增殖(Plt;0.05),增加前列腺特异抗原(Plt;0.05)及人体激肽释放酶2 mRNA表达(Plt;0.05)。结论EGF通过细胞内非受体酪氨酸激酶使AR特异位点磷酸化,诱导前列腺癌细胞增殖。
表皮生长因子;去势抵抗性前列腺癌; 雄激素受体;酪氨酸激酶;磷酸化
前列腺癌的发生及发展与雄激素有密切关系,通过外科去势(castration)或者药物从患者体内剔除雄激素是治疗前列腺癌的有效方法之一,但是, 患者最终对该治疗失去作用并逐渐进展为去势抵抗性前列腺癌(castration-resistant prostate cancer,CRPC),其机制尚不完全明了。有研究表明,低睾酮环境下雄激素受体(androgen receptor,AR)激活在CRPC发病中起关键作用[1-4]。在阉割动物,AR的过度表达具有促进前列腺癌移植瘤的形成作用,AR敲除后便抑制其肿瘤源性[2]。AR激活可能存在多种机理,包括AR基因扩增,AR过度表达,AR点突变,AR共激活因子的过度表达以及肿瘤自身产生的雄激素等[5]。近年来有研究发现非雄激素依赖的细胞内信号传导导致AR磷酸化是AR激活的重要途径之一[3-4,6]。表皮生长因子受体(epidermal growth factor receptor,EGFR)通过增加AR与性激素受体转录介导因子2(transcriptional intermediary factor 2,TIF2)的相互作用而增强AR的转录活性,人表皮生长因子受体2(human epidermal receptor 2,HER2)通过增加AR蛋白稳定性及与DNA结合力而激活AR的转录功能[7]。有研究报道在难治性前列腺癌中,表皮生长因子(epidermal growth factor,EGF)能够增加AR的活性[8]。本研究旨在探讨,在无雄激素的条件下,EGF对前列腺癌细胞增殖及雄激素受体磷酸化的影响。
1 材料与方法
1.1 试剂
EGF(Ramp;D公司); AR Tyr-267位点磷酸化特异性多克隆抗体及AR Tyr-534位点磷酸化特异性抗体(美国北卡罗来那大学综合肿瘤研究中心 Young E. Whang教授惠赠);鼠抗-AR单克隆抗体(Biogenex公司)。Ack1-siRNA、Src-siRNA及阴性对照scrambled siRNA(Dharmacon RNA Technologies)。
1.2 细胞及分组
雄激素依赖性前列腺癌细胞系LNCaP与LAPC4 (美国菌种保藏中心);分组为EGF处理组;DHT处理组;对照组。
1.3 AR磷酸化
在无雄激素环境下,EGF(100 ng/mL)处理LNCaP或LAPC-4细胞60 min,提取蛋白后Westernblot方法测定AR Tyr-267位点及Tyr-534位点磷酸化状态。
1.4 转染及基因敲除
利用转染试剂siPort Lipid(Ambion公司),将100 nmol/L Ack1-siRNA或Src-siRNA及阴性对照scrambled siRNA转染进入LNCaP或LAPC-4细胞,24 h后,给予EGF处理。
1.5 细胞增殖实验
将104个/孔的LNCaP或LAPC-4细胞培养于96孔培养板,给予二氢睾酮(dihydrotestosterone,DHT 10 nmol/L)或EGF(100 ng/mL)培养3 d,按照CCK-8说明书步骤检测细胞增殖情况。
1.6 定时定量RT-PCR
LNCaP及LAPC4细胞由EGF或DHT处理后,使用RNeasy Mini Kit(Qiagen公司)提取细胞总RNA,采用Taqman universal PCR体系(ABI)。引物及探针序列如下:PSAmRNA上游,5′-GGCAGCATT GAACCAGAGGAG-3′;PSA下游,5′-GCATGAACTTG GTCACCTTCTG-3′;PSA探针,5′-6-FAM-ATGACGTG TGTGCGCAAGTTCACCC-BHQ-1-3′;hK2上游,5′-GC CTTAGACCAGATGAAGACTCCA-3′;hK2下游,5′-GC CCAGGACCTTCACAACATC-3′;hK2探针,5′-6-FAM-TGACCTCATGCTGCTCCGCCTGT-BHQ-1-3′;GAPDH上游,5′-GTCATGGGTGTGAACCATGAGA-3′;GAPDH下游,5′-GGTCATGAGTCCTTCCACGATAC-3′;GAPDH探针,5′-6-FAM-CAGCCTCAAGATCATCAGCAATGC CTC-BHQ-1-3′。GAPDH为内参。
1.7 统计学分析
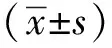
2 结果
2.1 EGF可诱导AR特异位点磷酸化
在无雄激素环境下,EGF可诱导LNCaP细胞及LAPC-4细胞AR Tyr-534和Tyr-267位点磷酸化(图1)。
2.2EGF通过两条不同的途径导致AR两个不同位点的磷酸化
通过siRNA转染方法敲除LNCaP细胞Src基因后,可有效抑制EGF诱导的AR Tyr-534位点磷酸化(图2A)。用同样方法敲除已知的引起AR Tyr-267位点磷酸化的Ack1基因[4],却不能抑制EGF诱导的AR Tyr-267位点磷酸化(图2B)。因此,EGF是通过两条不同的途径导致两个不同位点的磷酸化:1)通过细胞内Src激酶导致AR Tyr-534位点磷酸化;2)通过Ack1以外未知的细胞内激酶导致AR Tyr-267特异位点磷酸化。

图1 EGF诱导AR Tyr-534和Tyr-267位点磷酸化Fig 1 AR is phosphorylated at Tyr-267 and Tyr-534 after EGF stimulation
2.3在无雄激素的条件下,EGF能够促进前列腺癌细胞增殖
在无雄激素的条件下,EGF和DHT处理的LNCaP细胞吸光度值均明显高于对照组(Plt;0.01),LAPC-4细胞中也得到相似结果(图3)。
2.4EGF促进前列腺癌细胞PSA及hk2mRNA表达
EGF处理的LNCaP细胞PSAmRNA和hk2 mRNA表达水平均较对照明显上调(Plt;0.01),LAPC-4细胞中也得到相似结果(图4)。
3 讨论
近年来国外研究报道,低或无循环雄激素环境下前列腺癌细胞AR的磷酸化在CRPC发生中具有重要作用[3-4, 6, 9],而AR磷酸化机制尚不完全清楚。目前认为细胞外配体通过与前列腺癌细胞表面存在的EGF受体家族,包括EGFR,HER2等结合,通过细胞内非受体激酶信号传导最终导致AR磷酸化[1,4,6]。

A.Src knockdown inhibited AR phosphorylation at Tyr-534 induced by EGF; B.EGF-induced AR phosphorylation at Tyr-267 was not inhibited by Ack1 knockdown
图2SrcsiRNA沉默对EGF诱导的ARTyr-534及ARTyr-267磷酸化的影响
Fig2TheinfluenceofSrcknockdownonEGF-inducedARTyr-534phosphorylationandARTyr-267phosphorylation
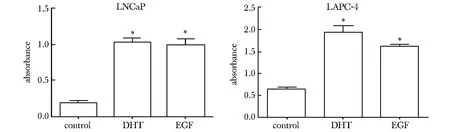
*Plt;0.05 compared with control图3 在无雄激素的条件下,EGF刺激雄激素依赖性前列腺癌细胞增殖

*Plt;0.01 compared with control图4 EGF促进前列腺癌细胞PSA及hK2 mRNA表达Fig 4 EGF increased PSA and hK2 mRNA expression
EGF可能通过Src激酶导致AR Try-534位点磷酸化[6];Heregulin与HER2结合通过细胞内Ack1酪氨酸激酶而导致前列腺癌细胞AR Try-267位点磷酸化[4]。本研究发现EGF可诱导AR Tyr-534和Tyr-267位点磷酸化,当Src敲除后,EGF诱导AR Tyr-534位点的磷酸化能够被抑制,说明Src激酶参与EGF下游信号通路,结果与报道[6]一致。然而,敲除引起AR Tyr-267位点磷酸化的Ack1基因,却不能抑制EGF诱导的AR Tyr-267位点磷酸化,说明EGF诱导的AR Try-267位点磷酸化与heregulin通过Ack1激酶导致的AR Try-267位点磷酸化存在不同通路[4],详细机制正在进一步研究中。
在无雄激素的条件下,EGF处理前列腺癌细胞后,仍可刺激细胞增殖,并且接近DHT刺激下细胞的增殖水平;前期也有报道,无雄激素条件下,heregulin[9]、bombesin[10]、白介素6[11]均可刺激前列腺癌细胞增殖,说明在无雄激素的条件下,非雄激素配体能够激活AR进而促进前列腺癌细胞增殖。AR靶基因PSA及hk2的表达可作为前列腺癌的肿瘤标志,与肿瘤负荷密切相关。无雄激素环境下,EGF处理的前列腺癌细胞的PSAmRNA及hk2 mRNA较对照显著增高。
本项研究发现EGF通过Src酪氨酸激酶和另一Ack1激酶以外的途径导致AR Tyr-534及Tyr-267位点磷酸化;在无雄激素存在环境下,促进前列腺癌细胞LNCaP及LAPC4增殖,增强前列腺癌细胞标志基因PSA及hk2 mRNA表达。结果提示EGF可能与CRPC发生有密切关系。通过动物实验及人体肿瘤标本的AR磷酸化研究将进一步验证本结果并将有助于最终揭示CRPC的发病机制。
[1] Lamont KR, Tindall DJ. Minireview: Alternative activation pathways for the androgen receptor in prostate cancer[J]. Mol Endocrinol, 2011, 25: 897-907.
[2] Chen CD, Welsbie DS, Tran C,etal. Molecular determinants of resistance to antiandrogen therapy[J]. Nat Med, 2004, 10: 33-39.
[3] Kraus S, Gioeli D, Vomastek T,etal. Receptor for activated C kinase 1 (RACK1) and Src regulate the tyrosine phosphorylation and function of the androgen receptor[J]. Cancer Res, 2006, 66: 11047-11054.
[4] Mahajan NP, Liu Y, Majumder S,etal. Activated Cdc42-associated kinase Ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation[J]. Proc Natl Acad Sci U S A, 2007, 104: 8438-8443.
[5] Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis[J]. J Clin Oncol, 2005, 23: 8253-8261.
[6] Guo Z, Dai B, Jiang T,etal. Regulation of androgen receptor activity by tyrosine phosphorylation[J]. Cancer Cell, 2006, 10: 309-319.
[7] Liu Y, Majumder S, Mccall W,etal. Inhibition of HER-2/neu kinase impairs androgen receptor recruitment to the androgen responsive enhancer[J]. Cancer Res, 2005, 65: 3404-3409.
[8] Gregory CW, Fei X, Ponguta LA,etal. Epidermal growth factor increases coactivation of the androgen receptor in recurrent prostate cancer[J]. J Biol Chem, 2004, 279: 7119-7130.
[9] Gregory CW, Whang YE, Mccall W,etal. Heregulin-induced activation of HER2 and HER3 increases androgen receptor transactivation and CWR-R1 human recurrent prostate cancer cell growth[J]. Clin Cancer Res, 2005, 11: 1704-1712.
[10] Lee LF, Guan J, Qiu Y,etal. Neuropeptide-induced androgen independence in prostate cancer cells: roles of nonreceptor tyrosine kinases Etk/Bmx, Src, and focal adhesion kinase[J]. Mol Cell Biol, 2001, 21: 8385-8397.
[11] Ueda T, Bruchovsky N, Sadar MD. Activation of the androgen receptor N-terminal domain by interleukin-6 via MAPK and STAT3 signal transduction pathways[J]. J Biol Chem, 2002, 277: 7076-7085.
Proliferation and androgen receptor phosphorylation of prostate cancer cells induced by epidermal growth factor at the absence of androgen
SUN Xue-fei1#, ZHAO Hui2#, LI Yang1, QIAN Jun1, BAI Xue-yan1, ZHOU Yong-jian3,ZHU Hong1, ZHANG Yong3, LIU Yuan-bo1,2*
(1.Dept. of Hematology and Oncology; 2.Dept. of Blood Transfusion; 3.Dept. of Urological Surgery, Beijing TianTan Hospital,Capital Medical University, Beijing 100050, China)
ObjectiveTo investigate the effect of epidermal growth factor on the proliferation and androgen receptor phosphorylation of prostate cancer cells in the absence of androgen.MethodsLNCaP and LAPC4 AR were used for the study, after EGF treatment,Western blot was used to detect AR phosphorylation in LNCaP and LAPC4 cells; Src and Ack1 genes with siRNA transfection were knocked down to observe the effect on androgen receptor phosphorylation; Cell Counting Kit-8(CCK-8) was used to measure the cell proliferation; Quantitive RT-PCR was used to analyze the expression of prostate-specific antigen and human kallikrein-2 mRNA.ResultsEGF can induce AR specific site phosphorylation at Try-534 and Try-267 by Src kinase and another unknown kinase; Compared with untreated control, EGF enharced the proliferation of prostate cancer cells(Plt;0.05) and increase the expression of prostate-specific antigen(Plt;0.05) and human kallikrein-2(Plt;0.05).ConclusionsEGF can induce proliferation and AR phosphorylation of prostate cancer by intracellular non-receptor tyrosine kinases
epidermal growth factor; castration-resistant prostate cancer; androgen receptor; tyrosine kinase;phosphorylation
2013-07-05
2013-09-26
国家自然科学基金(81272842,81302594)
*通信作者(correspondingauthor): ybliu1955@163.com
#对本文有相同贡献
1001-6325(2014)03-0305-05
研究论文
R 737.25
A
猜你喜欢
现代仪器与医疗(2021年6期)2022-01-18
天津医科大学学报(2021年3期)2021-07-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
世界科学技术-中医药现代化(2021年12期)2021-04-19
天津医科大学学报(2019年3期)2019-08-13
中国生殖健康(2019年7期)2019-01-06
中国男科学杂志(2016年5期)2016-12-01
中国男科学杂志(2016年5期)2016-12-01
中国医药生物技术(2015年4期)2015-12-26
川北医学院学报(2015年5期)2015-12-05
