
生产超低硫柴油的加氢脱硫催化剂级配技术
2014-11-18 08:23方向晨刘继华宋永一
化学反应工程与工艺 2014年5期
方向晨,郭 蓉,刘继华,宋永一
(1.中国石油化工股份有限公司抚顺石油化工研究院,辽宁 抚顺 113001;2.华东理工大学化学工程联合国家重点实验室,上海 200237)
柴油超深度加氢脱硫(ULDS)催化剂是目前炼油技术的开发热点[1-3],由于柴油加氢脱硫反应受硫化物类型、反应条件和原料性质(如芳烃、有机氮化物含量)等因素的影响,使得催化剂的应用选择复杂化,因此合理选择催化剂成为相关加氢脱硫技术研究工作的重点内容之一。
先进炼油技术公司(ART)开发了SmART Catalyst SystemTM级配技术,即常规的Mo-Ni 和Mo-Co型催化剂的级配装填,其目的主要是追求在较低氢耗下达到满意的脱硫效果[4,5]。Albemarle 公司开发了STAX 级配装填技术,即考虑到高加氢活性的Nebula 催化剂氢耗高和制造成本高的缺陷,为了寻求在超深度脱硫活性、装置氢耗及催化剂费用三方面的平衡点,在不同压力下考察了Nebula 催化剂和Mo-Co 型KF-767 及Mo-Ni 型KF-848 等催化剂组合使用时,装填在反应器不同区域对精制柴油硫含量的影响。研究表明,即使是在不受热力学平衡限制的反应温度条件下,不同压力等级的加氢装置,高加氢活性Nebula 催化剂装填位置不同,对柴油中硫含量的脱除效果也不相同[6-8]。但是,在受热力学平衡限制条件下如何选择更合理的催化剂级配体系具有重要意义。
硫化物的加氢脱硫过程一般有三种反应途径:加氢然后氢解脱硫途径(加氢途径)、直接氢解脱硫途径(氢解途径)和烷基转移然后氢解脱硫途径(烷基转移途径)。在柴油超深度加氢脱硫的条件下,关键是要脱除柴油中有空间位阻效应的4,6-二甲基二苯并噻吩(4,6-DMDBT)类硫化物。这类硫化物的脱除主要通过加氢途径和烷基转移途径完成。目前大多数商业催化剂主要依靠的是提高催化剂的加氢途径。最近,Fang 等发现了具有良好烷基转移反应活性的加氢脱硫催化剂[3],特别是在反应温度需要较高的应用场合,能够不受芳烃饱和热力学平衡的限制实现超深度加氢脱硫,且表现出了催化剂高温活性稳定性好、对硫化氢的影响不太敏感等突出优点。
为了更好地发挥不同类型催化剂的优势,本工作就Fang 等[3]开发的W-Mo-Ni 催化剂(加氢途径反应活性好的催化剂)和Mo-Co 催化剂(具有烷基转移反应活性的催化剂),针对反应器不同床层在反应过程中工况条件和原料性质特点,结合不同类型催化剂在不同条件下超深度脱硫时的优缺点,考察不同类型催化剂级配装填的反应效果,以期指导工业装置优化应用超深度加氢脱硫催化剂。
1 实验方法
实验装置由液体原料,氢气供应模块、反应器模块、反应后气液分离模块和气体净化循环模块组成。催化剂装填量为100 mL,采用惰性石英砂稀释,稀释比为3:1。单种催化剂装填于一个反应器中,两种催化剂级配采用同样稀释比后装填于串联的两个反应器中(R1 和R2),体积比为1:1。实验用氢气为经过高压加氢脱氧及硅胶/分子筛脱水净化处理后的电解氢,氢纯度大于99.9%(V)。
以直馏柴油(HSRGO)、焦化柴油(DCD)、催化柴油(LCO)、1#和2#混合柴油为原料(性质见表1),在反应氢分压6.4 MPa、氢油体积比400/1、体积空速1.5 h-1等反应条件下,考察了W-Mo-Ni和Mo-Co 两种类型催化剂以及两种催化剂不同级配方式下的活性对比。
通过分析反应后的液体产品中硫、氮含量来表征催化剂活性,采用紫外荧光法测定产物中硫含量(HS/T 0689-2000),采用化学发光法测定产物中氮含量(HS/T 0657-2007)。
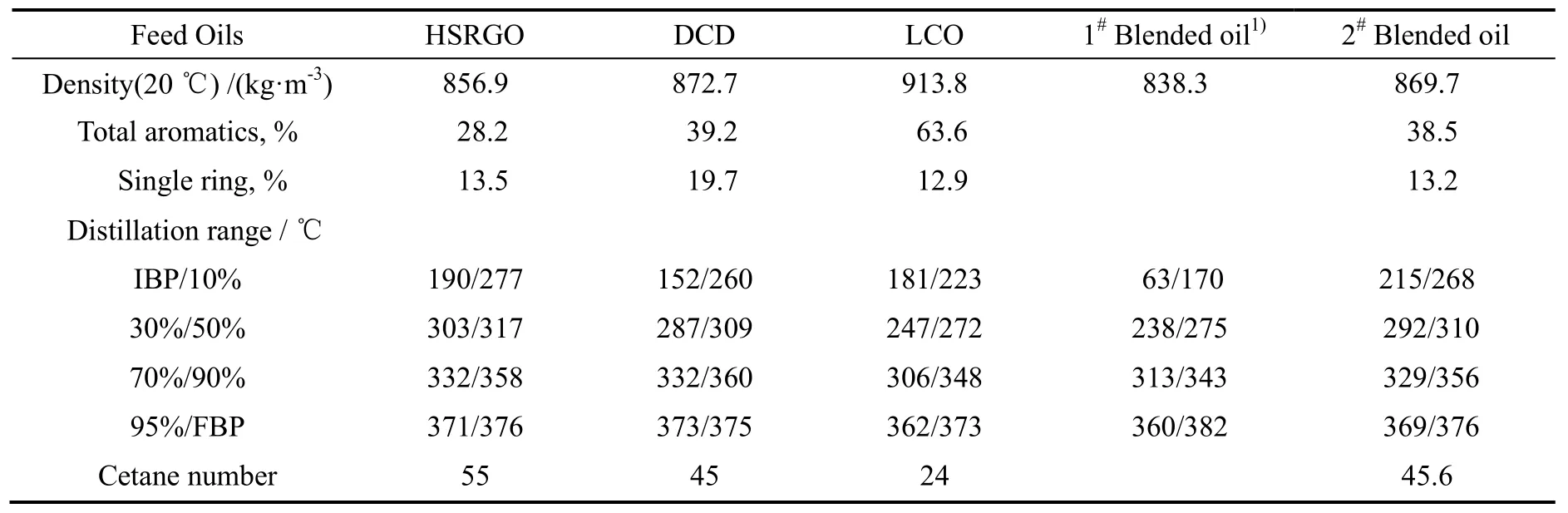
表1 不同柴油原料性质Table 1 The properties of different diesel feeds
2 结果分析与讨论
两种不同类型催化剂以及两种催化剂不同级配方式下的活性对比结果见表2~表6。

表2 不同催化剂及级配方式加工HSRGO 时的超深度脱硫效果Table 2 Results for different catalysts and stacking when treating HSRGO
由表2可以看出,由于直馏柴油中的氮和芳烃含量低,因而氮和芳烃对超深度脱硫的影响相对较小,催化剂的主要任务是完成加氢脱硫反应,因此单独的Mo-Co 型催化剂体现出更好的脱硫活性,对于催化剂级配,则W-Mo-Ni/Mo-Co 的脱硫效果优于Mo-Co/W-Mo-Ni。
对于氮含量非常高的焦化柴油DCD,单独使用加氢性能好的W-Mo-Ni 型催化剂具有较好的超深度脱硫效果(见表3),其脱硫活性的由大到小为:W-Mo-Ni,Mo-Co/W-Mo-Ni,W-Mo-Ni/Mo-Co 和Mo-Co。这是由于柴油加氢脱硫反应中,有机氮化物是催化剂的活性抑制剂[9],而W-Mo-Ni 催化剂的加氢脱氮活性要明显高于Mo-Co 催化剂,从而够降低有机氮化物对加氢脱硫反应活性的抑制作用。
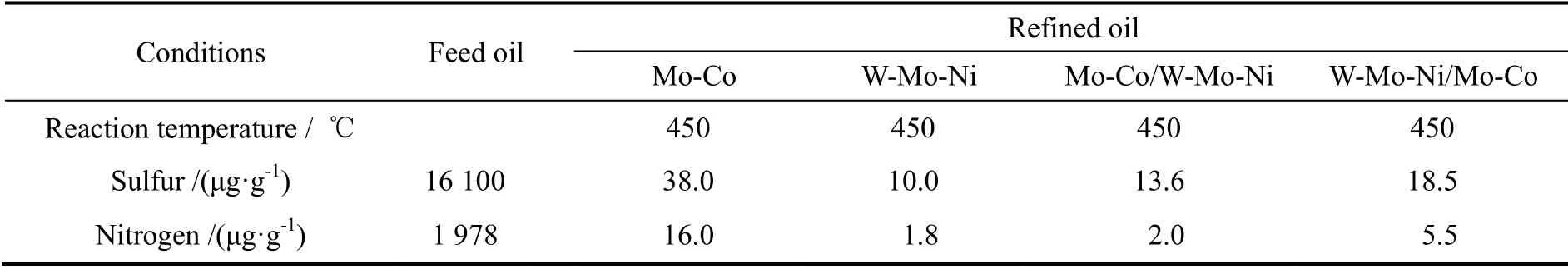
表3 不同催化剂及级配方式加工DCD 时的超深度脱硫效果Table 3 Results for different catalysts and stacking when treating DCD
由表4可知,对于硫、氮及芳烃含量高、密度大的LCO,加氢性能好的W-Mo-Ni 催化剂反应温度比Mo-Co 型催化剂低10 ℃。这是由于LCO 中多环芳烃含量高,对应地原料中所含的4,6-二甲基二苯并噻吩类硫化物含量也高,脱硫反应受催化剂芳烃饱和性能影响大,因而加氢性能较好的催化剂体现出更好的超深度脱硫活性。与处理直馏柴油、焦化柴油结果比较,虽然反应温度高了10~20 ℃,但产品的硫含量也远达不到要求(低于10 µg/g)。

表4 不同催化剂加工LCO 时的超深度脱硫效果Table 4 Results for different catalysts when treating LCO
图1为反应温度对催化柴油该柴油原料加氢脱硫反应的影响。从图1可以看出,对W-Mo-Ni 型催化剂加氢脱硫转化率先随着温度的升高而升高,继续升高温度加氢脱硫转化率则随着温度的升高而降低,这一趋势与柴油产品中芳烃加氢转化率随温度的变化趋势正好呈对应关系。
表5为1#blended oil 的加氢脱硫实验结果。可见W-Mo-Ni/Mo-Co 加氢脱硫活性最好,W-Mo-Ni 和Mo-Co 次之,Mo-Co/W-Mo-Ni 活性最弱。这是由于柴油原料的硫、氮含量较高,而芳烃含量不高因此W-Mo-Ni/Mo-Co 的催化剂级配方式可以利用W-Mo-Ni 催化剂先解除有机氮化物对Mo-Co 催化剂活性的抑制作用,发挥Mo-Co 催化剂在低氮条件下加氢脱硫反应活性高的优势(这一点对照表中直馏柴油结果可以看得更清楚)。反过来级配则两种催化剂都不能在最有利的条件下操作,从而产生了负效应。
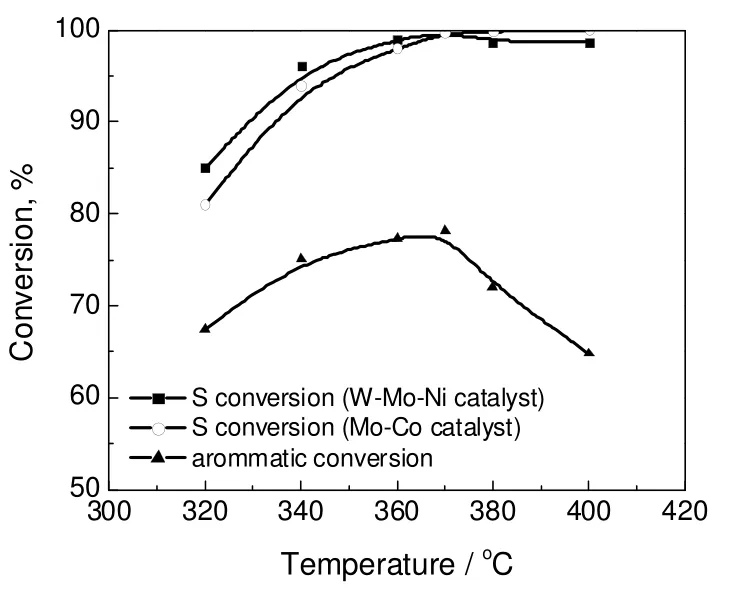
图1 反应温度对加氢脱硫(HDS)和加氢脱芳(HAD)的影响Fig.1 Effects of reaction temperature on HDS and HAD
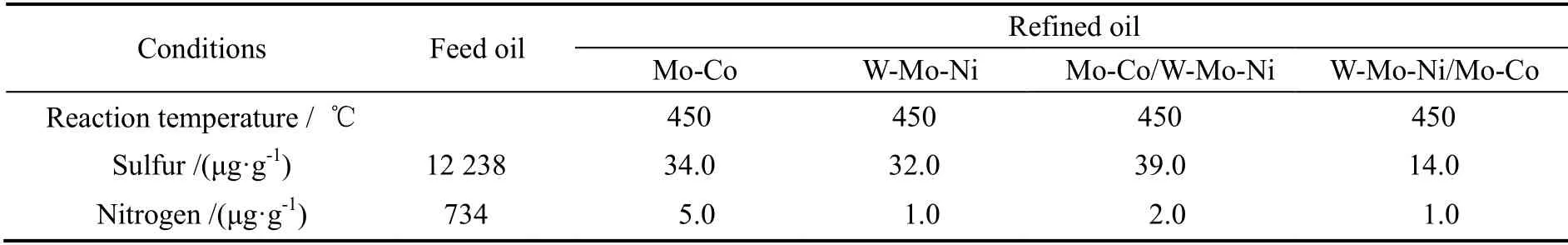
表5 不同催化剂及级配方式加工1#blended oil 时的超深度脱硫效果Table 5 Results for different catalysts and stacking when treating 1#blended oil
表6为2#blended oil 在两个串联反应器中的加氢脱硫实验结果,2#blended oil 是由70%高干点直馏柴油与30%催化柴油混合而成,其原料性质与1#blended oil 基本相似。实验主要模拟工业装置控制不同催化剂床层反应温度,考察了不同催化剂级配方式对加氢脱硫反应结果的影响。由于二反应温度均比一反温度高20 ℃,而一反出来的精制油品中W-Mo-Ni/Mo-Co 级配方式的脱硫效果不如Mo-Co/W-Mo-Ni 级配方式,但W-Mo-Ni/Mo-Co 级配方式的综合脱硫效果好于Mo-Co/W-Mo-Ni 级配方式,且反应温度越高,优势越明显。这是由于W-Mo-Ni 催化剂在一反能够更好的脱除有机氮化物和芳烃,为Mo-Co 催化剂作用的发挥提供了更好的条件;由表6还可看出,在较高温度下,芳烃加氢饱和将受到热力学平衡限制,W-Mo-Ni 催化剂放在二反高温下操作会产生负效应,而将具有烷基转移功能的Mo-Co 型催化剂放在二反高温下操作,使这类催化剂在高温下更加有利于烷基转移脱硫功能的发挥,从而更好地实现超深度脱硫的目标。由图1可以看出,与W-Mo-Ni 催化剂相比,提高反应温度对提高Mo-Co 催化剂一直有效,表明该催化剂通过烷基转移途径实现超深度加氢脱硫,避免了高温下加氢途径受芳烃饱和热力学平衡影响的问题。

表6 不同催化剂级配方式加工2#blended oil 时的深度脱硫效果Table 6 Results for different catalysts grading when treating 2#blended oil
综上所述,对于氮及多环芳烃含量低、十六烷值高的直馏柴油,应选择直接脱硫活性好的Mo-Co型催化剂;对于氮含量和/或芳烃含量非常高的焦化柴油及催化柴油类二次加工柴油,应选择加氢性能好的W-Mo-Ni 类催化剂;对于直馏柴油搀兑部分二次加工柴油的混合油,采用W-Mo-Ni/Mo-Co 级配方式比采用单一品种催化剂具有更好的超深度脱硫效果。
3 超深度加氢脱硫条件下柴油加氢脱硫反应动力学模型
实验结果表明,为工业装置确定优化的超深度加氢脱硫反应条件,选择最合适的催化剂或催化剂级配体系是一种复杂的多变量系统评估过程,建立相应的加氢脱硫反应动力学模型能为这一优化过程提供更好的数值分析工具。馏分油的加氢脱硫反应动力学研究[9]较多,但目前对超深度脱硫反应动力学研究多集中在对机理反应的探讨上,本工作尝试着开发一种能够指导工业应用的柴油超深度加氢脱硫反应动力学模型并取得了初步的有益结果。
柴油馏分中的含硫化合物根据其加氢脱硫反应难易程度大致可分为两类:一类是非噻吩类硫化物、噻吩类硫化物以及含有一个或两个芳环的苯并噻吩类硫化物,主要包括硫醇、硫醚以及噻吩(T)、苯并噻吩(BT)和二苯并噻吩(DBT)等,这类硫化物沸点较低,主要存在于中低沸点石油馏分中,比较容易被脱除;另一类是含有多个芳环,并且芳环上有取代基的高沸点的多苯并噻吩类硫化物,主要包括4,6-二甲基二苯并噻吩、2,4,6-三甲基二苯并噻吩等,这类物质沸点较高,结构稳定,不容易被脱除[10]。加氢脱硫反应的活性主要取决于硫化合物分子的大小及结构。一般来说,对于同一结构的硫化物,分子越大,HDS 的反应速率就越低,但其影响程度远低于分子结构的影响[11]。
将柴油分成2 个集总,硫醇、硫醚、噻吩类硫化物等分为1 个集总,称为I 型硫;二苯并噻酚,特别是4,6 位有取代基的4,6-DMDBT 类硫化物划为第2 集总称为II 型硫。这两类硫的量可由GC-AED硫结构分析仪得到。I 型硫脱除是按氢解途径进行,其反应级数对硫化物为一级。II 型硫是按氢解途径、加氢途径和烷基转移途径同时进行反应,且所有反应的级数(包括脱氢反应)对硫化物均为一级。其反应网络示意图如图2所示,其中B 为加氢中间体,E 是烷基转移路径中间体,所以产品最后总硫:CS=CSI+CSII+CB+CE。I 型硫的初始浓度C10,II 型硫的初始浓度C20。
根据上述模型,可得:
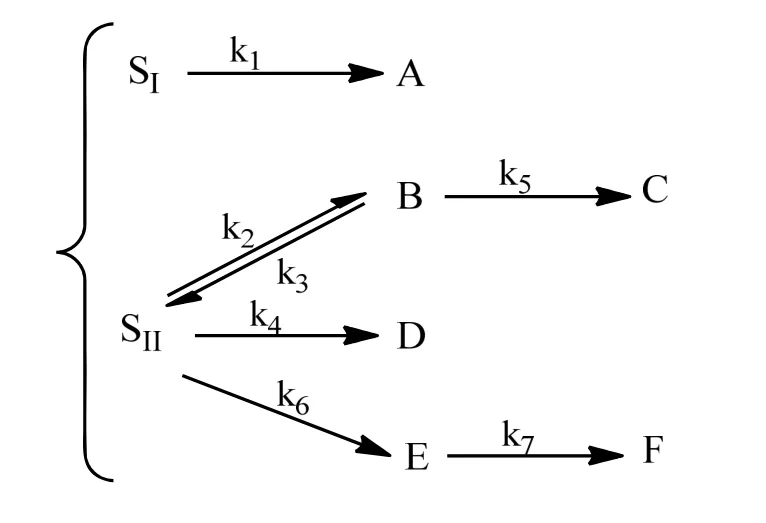
图2 柴油超深度加氢脱硫二集总三反应途径网络Fig.2 The structure of two lumps and three reaction routes for ULDS

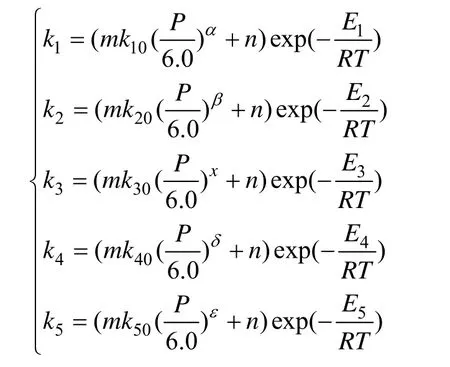
其中,m和n是原料馏程(中平均沸点温度)和密度对反应速度影响的校正系数[9],如下式:

式中Tavg.boiling(base-oil)、Tavg.boiling(model-oil)分别为原料油及参比原料油的中平均沸点,APIbase-oil、APImodel-oil分别为原料油和参比原料油的API。
有机氮和硫化氢对加氢脱硫反应的影响均按对相应反应的吸附中毒模型考虑[9]。该模型对Mo-Co催化剂的回归计算结果如表7所示,14 个数据的残差平方和仅为0.987;该模型应用于预测本研究第二部分所列实验数据的结果如图3所示,预测的平均相对误差(对产品硫含量)为11%,最大相对误差为31%。可以看出该模型基本满足应用的要求,它也是目前能够比较全面地反映原料性质、反应条件和催化反应路径对柴油超深度加氢脱硫反应影响的动力学模型。

图3 模型预测结果与实验结果偏差对比Fig.3 The deviation comparasou between model calculated and tested

表7 HSRGO 实验值与模型计算值的对比Table 7 Comparison between experiment data and calculated data by model of HSRGO

续表7
4 催化剂级配技术的工业应用结果
上海石化3.3×106t/a 柴油加氢装置以直柴加40%的焦化汽柴油和催化柴油的混合柴油为原料,硫氮含量较高,设计空速高达2.3 h-1。该装置采用W-Mo-Ni/Mo-Co 催化剂级配技术。该装置于2010年6月开工,于8月底进行了高空速条件下生产国IV 标准清洁柴油的活性标定,标定结果见表8。
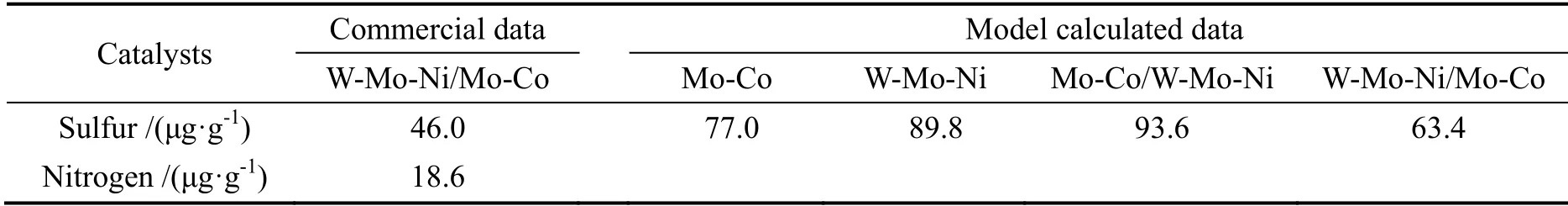
表8 上海石油化工股份有限公司3.3×106 t/a 柴油加氢装置工业应用结果Table 8 The commercial results in Shanghai Petrochemical Company Limited 3.3×106 t/a
表8给出了动力学模型计算结果,其反应温度按照反应器二床层温度高于一床层温度20 ℃来考虑,这是由于所建模型未考虑反应热对反应床层温升的影响,因此在计算过程中对每个床层按等温反应器考虑。虽然计算的结果与实际结果相差37%,但仍可反映出在这种原料和装置条件下,W-Mo-Ni/Mo-Co 催化剂级配体系的反应效果要好于单独使用W-Mo-Ni、Mo-Co 催化剂和使用Mo-Co/W-Mo-Ni 催化剂级配体系。
级配技术在该装置上目前已运行38 个月。从长周期生产国IV 标准柴油的反应条件看,2010年8月标定期间生产国IV 标准柴油时反应器入口温度为320 ℃,至2012年3月,在反应压力、体积空速及原料油性质基本相同的条件下生产国IV 标准柴油时,其反应器入口温度为330 ℃,19 个月温度只提高10 ℃,提温速率为6.316 ℃/a,体现出催化剂级配体系很好的活性稳定性及对原料油良好的适应性,为上海地区柴油产品质量升级提供了有力的技术支撑。
5 结 论
柴油的超深度加氢脱硫是一个复杂的反应体系。对于氮及多环芳烃含量低、十六烷值高的直馏柴油,应选择直接脱硫活性好的Mo-Co 型催化剂;对于氮含量和/或芳烃含量非常高的焦化柴油及催化柴油类二次加工柴油,应选择加氢性能好的W-Mo-Ni 类催化剂;对于直馏柴油搀兑部分二次加工柴油的混合油,采用W-Mo-Ni/Mo-Co 级配方式比采用单一一种催化剂具有更好的超深度脱硫效果。
根据复杂硫化物的多途径加氢脱硫反应实际,建立了一个能够较全面反映原料性质、反应条件和催化反应路径对柴油超深度加氢脱硫反应影响的柴油超深度加氢脱硫反应动力学模型。该模型的建立能够为加氢脱硫催化剂级配技术的应用提供分析计算的基础。
催化剂级配技术加工硫氮含量较高的直馏柴油与二次加工油品混合原料油时,取得了非常好的工业应用效果,且活性稳定性提高、原料适应性增强。
[1]Stanislaus A, Marafi A, Rana M S.Recent advances in the science and technology of ultra-low sulfur diesel (ULSD) production [J].Catalyst Today, 2010, 153(1-2):1-68.
[2]Shafi R, Hutchings G J.Hydrodesulfurization of hindered dibenzothiophenes:an overview [J].Catalyst Today 2000, 59(3-4):423-442.
[3]Fang X C, Guo R, Yang C M.The development and application of catalysts for ultra-deep hydrodesulfurization of diesel [J].Chinese Journal Catalyst, 2013, 34(1):130-139.
[4]Charles W, Olsen Ph D, David K.Custom catalyst systems for meeting ULSD regulations [C].NPRA Annual Meeting, AM-05-17, San Francisco, USA, 2005.
[5]Charles W O, Gerianne D’Angelo.No need to trade ULSD catalyst performance for hydrogen limits:SmART approaches [C]NPRA Annual Meeting, AM-06-06, Salt Lake City, USA, 2006.
[6]Mayo S, Leliveld B.Experiences in maximizing performance of ULSD units [C].NPRA Annual Meeting, AM-09-14, San Antonio, USA,2009.
[7]Mayo S, Vogt K, Leliveld B, et al.Crossing frontiers in the performance and economic return of ULSD units [C].NPRA Annual Meeting, AM-10-170, Phoenix, Arizona, USA, 2010..
[8]Mayo S.Successful production of ULSD in low pressure hydrotreaters [C].NPRA annual Meeting, AM-11-23, San Antonio, USA, 2011.
[9]方 星, 方向晨, 程振民, 等.柴油加氢脱硫反应动力学研究 [J].当代化工, 2005, 34(3):145-148.Fang Xing, Fang Xiangchen, Cheng Zhenmin, et al.Reaction kinetics of diesel deep hydrodesulfurization [J].Contemporary Chemical Industry, 2005, 34(3):145-148.
[10]Da Costa P, Potvin C, Manoli J M, et al.New catalysts for deep hydrotreatment of diesel fuel kinetics of 4,6-dimethyldibenzothiophene hydrodesulfurization over alumina-supported molybdenum carbide [J].Journal of Molecular Catalysis A:Chemical, 2002,184(1-2):323-333.
[11]Ksbe T, Aoyama Y, Wang D H, et al.Effects of H2S on hydrodesulfurization of dibenzothiophene and 4,6-dimethyldibenzothiophene on alumina-supported NiMo and NiW catalysts [J].Applied Catalyst A:General, 2001, 209(1-2):237-247.
猜你喜欢
科学家(2021年24期)2021-04-25
当代化工研究(2016年1期)2016-03-16
中国资源综合利用(2016年7期)2016-02-03
合成化学(2015年10期)2016-01-17
环境科技(2015年3期)2015-11-08
橡胶工业(2015年9期)2015-08-29
橡胶工业(2015年6期)2015-07-29
橡胶工业(2015年4期)2015-07-29
电源技术(2015年9期)2015-06-05
应用化工(2014年11期)2014-08-16
