
黄芩素结构修饰的研究
2014-11-09 01:22侯珏卓
吉林化工学院学报 2014年5期
侯珏卓,陈 喆
(天津药物研究院药业有限责任公司技术开发部,河北天津300193)
黄芩素(Baicalein)是传统中药黄芩(Scutellaris baicalensis georgi)的有效成分黄芩苷(Baicalin)的苷元,具有抗氧化[1]、抗菌、抗病毒[2]、抗过敏[3]、抗炎、降压[4]、利尿、利胆[5]等多种生理活性,临床上主要用于抗菌消炎和抗感染.目前,有关黄芩素的制剂已在临床上应用于上呼吸道感染、急性扁桃体炎、咽炎、慢性阻塞性肺病,传染性肝炎、急慢性胃肠炎及细菌性痢疾、肾盂肾炎等,显示出了较好的疗效.
黄芩素具有良好的药理活性,毒副作用微小,而且在我国资源丰富,来源广泛,经济成本低,但是黄芩素结构中存在3个相邻酚羟基,易于形成分子内氢键,造成其亲脂性、亲水性均较差,黄芩素类药物在临床应用上存在着溶解性差、生物利用度低等缺点[6],制约着其在现代中药建设中的发展.近几年,如何提高黄芩素类药物的溶解性与生物利用度,增加在人体的吸收,成为了国内外的研究热点.目前关于黄芩素活性方面的研究报道较多[7],但是对黄芩素结构修饰及构效关系的研究报道甚少.由于黄芩素的结构中A环有羟基取代,可作为先导化合物进行结构修饰以发掘这类化合物潜在的生理功能.故本研究以黄芩素为母体,根据黄酮类化合物的构效关系,通过化学的方法改变其结构或引入能转变成盐的官能团,合成了一系列黄芩素类衍生物,实现增加溶解性的目的,是一种药物创新的有效途径[8].
1 实验部分
1.1 仪器与试剂
RE-52AA旋转蒸发仪(上海亚荣生化仪器厂);90-3恒温双向磁力搅拌器(上海振荣科学仪器有限公司);瑞士Bruker Avance500核磁共振波谱仪(DMSO-D6或 CDCl3-d为溶剂,TMS为内标).
黄芩素(实验室提取分离纯化);丙酮、醋酐、石油醚、氯仿(天津市江天化工技术有限公司);无水碳酸钾、甲醛水溶液(天津市百世化工有限公司);溴苄(天津精益合成科技有限公司);苄胺、1,4-二溴丁烷(阿法埃莎化学有限公司);氮气(天津市兴盛气体有限公司);硅胶(上邦实业天津分公司);实验室所用的试剂与溶剂均为分析纯.
1.2 化合物的制备
黄芩素按文献[9]方法制备,利用萃取、柱层析等分离、纯化方法从中药黄芩的甲醇提取液中分离出黄芩素粗品,利用重结晶等纯化方法进一步得到黄芩素晶体.以黄芩素为母体,按图1所示研究路线,合成5种黄芩素类衍生物.
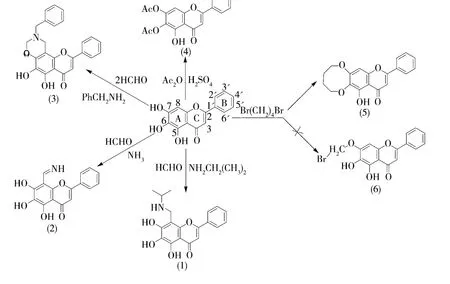
图1 黄芩素的结构修饰
1.2.1 黄芪素衍生物1的合成
8-(N-异丙胺基)-亚甲基-5,6,7-三羟基-2-苯基-4H-1-苯并吡喃-4-酮(黄芪素衍生物1)的合成:称量黄芩素0.1 g于50 mL三颈烧瓶中,加入甲醇10 mL和甲醛水溶液(w/w=36%)0.03 mL,升温至65℃,至反应液澄清.缓慢加入异丙胺0.047 mL,加热回流搅拌2 h后,旋蒸除去溶剂,得黄色固体粗品.将得到的黄色固体进一步上反相柱纯化,得到淡黄色固体,精品收率75%.
1.2.2 黄芪素衍生物2的合成
8-烯胺甲基-5,6,7-三羟基-2-苯基-4H-1-苯并吡喃-4-酮(黄芪素衍生物2)的合成:称量黄芩素0.1 g于50 mL三颈烧瓶中,加入甲醇10 mL和甲醛水溶液(w/w=36%)0.03 mL,升温至65℃,至反应液澄清.缓慢加入氨水(w/w=35.28%)0.11 mL,加热回流反应30 min.旋蒸除去溶剂,得黄色固体粗品.将得到的黄色固体进一步上反相柱纯化,得到淡黄色固体,精品收率86%.
1.2.3 黄芪素衍生物3的合成
二氢苯并噁嗪类化合物(黄芪素衍生物3)的合成:称量黄芩素0.1 g于50 mL三颈烧瓶中,加入甲醇10 mL和甲醛水溶液(w/w=36%)0.03 mL.缓慢滴加苄胺0.05 mL,加热回流反应3h.旋蒸除去溶剂,得黄色固体粗品.将得到的黄色固体进一步上反相柱纯化,得到淡黄色固体,精品收率69%.
1.2.4 黄芪素衍生物4的合成
5-羟基-6,7-二乙酰氧基-2-苯基-4H-1-苯并吡喃-4-酮(黄芪素衍生物4)的合成:称量黄芩素0.27 g与醋酐5 mL置于50 mL三颈圆底烧瓶中,缓慢滴加浓硫酸6滴,加热搅拌反应30 min,自然降温至室温.将反应液倾倒入已经冰浴的蒸馏水50 mL中,用5%碳酸氢钠调节溶液pH值至弱碱性,水相用乙酸乙酯30 mL萃取3次.合并乙酸乙酯层.加入无水硫酸镁干燥,静置过夜,真空蒸除溶剂,过硅胶柱,洗脱剂:V(石油醚)V(丙酮)=9 2,得淡黄色固体,收率80%.1.2.5 黄芪素衍生物5的合成
5-羟基-6,7-正丁二氧基-2-苯基-4H-1-苯并吡喃-4-酮(黄芪素衍生物5)的合成:称量黄芩素0.14 g置于50 mL三颈烧瓶中,加入30 mL丙酮溶解.氮气保护下依次加入0.08 mL1,4-二溴丁烷,0.2 g无水碳酸钾,0.2 g碘化钠,加热回流搅拌48 h后,过滤,在滤液中加入几滴醋酸,真空旋蒸除去丙酮后,残留物丙酮重结晶,得到浅黄色固体,精品收率85%.
1.3 化合物的结构表征
利用1H-NMR对所制备的黄芩素及合成的五种化合物做如下表征.
1.3.1 黄芩素的结构表征
1H NMR(500MHz,DMSO-D6),δ:12.65(1H,s,5-OH),10.56(1H,s,7-OH),8.80(1H,s,6-OH),8.05-8.08(2H,m,-C6H5),7.54-7.61(3H,m,-C6H5),6.92(1H,s,3-H),6.62(1H,s,8-H).
1.3.2 黄芩素衍生物1的结构表征
1H NMR(300MHz,DMSO-D6),δ:12.45(1H,s,5-OH),7.99-8.02(2H,m,-C6H5),7.55-7.56(3H,m,-C6H5),6.71(1H,s,3-H),4.26(2H,s,N—CH2-),3.32(1H,m,N-CH),1.28-1.29(6H,s,-2CH3).
1.3.3 黄芩素衍生物2的结构表征
1H NMR(300MHz,DMSO-D6),δ:12.85(1H,m,5-OH),7.82-7.84(2H,m,-C6H5),7.44-7.45(3H,m,-C6H5),6.86(1H,s,3-H),4.40(1H,s,N=CH-).
1.3.4 黄芩素衍生物3的结构表征
1H NMR(300MHz,CDCl3-d),δ:12.45(1H,s,5-OH),7.76-7.78(2H,m,-C6H5),7.49-7.54(3H,m,-C6H5),7.34-7.38(5H,m,-C6H5),6.68(1H,s,3-H),5.08(2H,s,-O-CH2-N),4.26(2H,s,Ar-CH2-),3.97(2H,s,-C-CH2-N-).
1.3.5 黄芩素衍生物4的结构表征
1H NMR(300MHz,CDCl3-d),δ:12.95(1H,s,5-OH),7.88-7.90(2H,m,-C6H5),7.54-7.59(3H,m,-C6H5),6.98(1H,s,3-H),6.75(1H,s,8-H),2.36-2.38(6H,m,2CH3CO-).
1.3.6 黄芩素衍生物5的结构表征
1H NMR(300MHz,CDCl3-d),δ:12.78(1H,s,5-OH),7.86-7.88(2H,m,-C6H5),7.49-7.54(2H,m,-C6H5),6.63(1H,s,3-H),6.54(1H,s,8-H),4.74(2H,t,1″-H),4.24(2H,t,4″-H),2.06(2H,m,2″-H),1.83(2H,m,3″-H).
2 结果与讨论
在黄芩素1H-NMR中,黄酮母核A环上5位酚羟基易与相邻4位羰基发生分子内缔合形成氢键,如图2所示,有去屏蔽作用,出现在较低场,造成5位羟基化学位移大于6、7位羟基氢的化学位移,如图3所示,因此12.65处单峰归属为A环5位酚羟基的氢;黄酮母核A环上的羟基氢均出现在谱图中的低场,8.80、10.56处单峰分别归属为黄酮母核A环6位、7位羟基氢;由于B环2'位于6'位的氢处于C环双键的去屏蔽区,受到各向异性效应的影响,共振信号比其它质子出现在相对低场,因此8.05-8.08 处多重峰归属为B 环2'、6'位的的2个氢;7.54-7.61处多重峰归属为B环上3'、4'、5'位的3个氢;6.92处单峰归属为C环3位上的氢,6.62处单峰归属为A环8位上的氢.
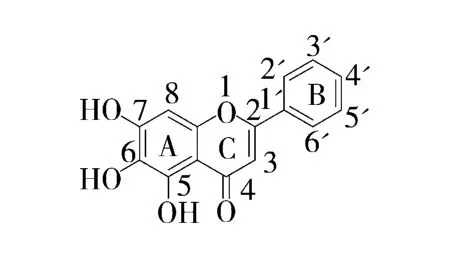
图2 黄芩素的化学结构
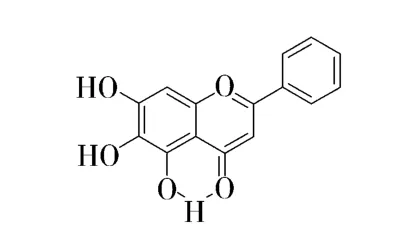
图3 黄芩素的氢键效应
2.1 黄芩素A环8位的结构修饰
黄芩素A环8-位氢与脂肪伯、仲胺或芳香伯、仲胺在甲醛水溶液中按照一定摩尔配比进行Mannich反应,产物可通过与酸反应生成盐类,达到增加其溶解性的目的.该反应条件温和,制备步骤简单,收率高.如果改变伯胺与甲醛的毫摩尔比后,得到含六元环的二氢苯并噁嗪衍生物(3).可能是反应产物Mannich碱进一步与甲醛反应生成N-取代羟甲基化中间体,再脱水环合,得到含六元环的二氢苯并噁嗪衍生物(3).故在与伯胺反应中要注重伯胺与甲醛比,以得到目标产物.
本文合成并利用1H-NMR鉴定以下3个Mannich碱衍生物.
2.1.1 黄芩素衍生物1的表征
对照黄芩素1H-NMR谱图,在化合物(1)的1H-NMR中,12.45处单峰归属为A环5位羟基氢;7.99-8.02处多重峰归属为B环2'位于6'位的氢;7.55-7.56 处多重峰归属为 B 环上3'、4'、5'位的3个氢;6.71处单峰归属为C环3位上的氢;4.26处单峰归属为A环8位亚甲基上的两个氢.
同时,区别于黄芩素1H-NMR谱图,化合物(1)1H-NMR谱图中在高场出现了两组新信号.将3.32处多重峰归属为A环8位侧链N-CH叔碳上的氢,1.28-1.29处单峰归属为A环8位侧链-HNCH(CH3)2两个甲基上的6个氢.因此,可以鉴定成功合成了化合物(1).
2.1.2 黄芩素衍生物2的表征
对照黄芩素1H-NMR谱图,在化合物(2)的1H-NMR中,12.85归属处单峰位为A环5位酚羟基,7.82-7.84 与7.44-7.45 归属为 B 环的5 个氢,6.86处单峰归属为C环3位上的氢.
同时,区别于黄芩素1H-NMR谱图,化合物(2)1H-NMR谱图中4.40处出现了一组新的单峰,归属为A环8位侧链NH=CH-上次甲基的氢.因此,可以鉴定成功合成了化合物(2).
2.1.3 黄芩素衍生物3的表征
对照黄芩素1H-NMR谱图,在化合物(3)的1H-NMR中,12.45处单峰归属为A环5位酚羟基,7.76-7.78 与7.49-7.54 处多重峰归属为 B 环的5个氢,6.68处单峰归属为C环3位上的氢.
同时,区别于黄芩素1H-NMR谱图,化合物(3)1H-NMR谱图中,出现了四组新的信号,7.34-7.38处多重峰归属为饱和六元环氮上N-CH2-Ph部分苯基上的5个芳香氢,5.08处单峰归属为饱和六元环-O-CH2-N部分亚甲基上的2个氢,4.26处单峰归属为饱和六元环Ph-CH2-部分亚甲基上的2个氢,3.97处单峰归属为饱和六元环-C-CH2-N-部分上的2个氢.因此,可以鉴定成功合成了化合物(3).
通过Mannich反应在黄芩素8位α氢引入了胺甲基类基团,成功合成并鉴定了3个Mannich碱衍生物.通过引入胺甲基对天然药物活性成分黄芩素进行结构修饰,利用胺甲基成盐能够改善此类药物的水溶性,不仅促进了此类药物在人体内的转运过程,同时也提高了其在作用部位的有效浓度,进而改善了药物的吸收性能,提高药物生物利用度,是一种提高黄芩素类化合物水溶性的有效方法.
2.2 黄芩素C环6、7位的结构修饰
利用乙酰基保护6,7位羟基成功合成了化合物(4).同时利用黄芩素的7-位羟基的较强酸性选择性的与溴取代物反应,脱去溴化氢.但通过1H-NMR鉴定,没有得到预期产物(6),而是生成了环状化合物(5),如图1所示.反应说明黄芩素的6,7位羟基的反应选择性不高.2.2.1 黄芩素衍生物4的表征
对照黄芩素1H-NMR谱图,在化合物(4)的1H-NMR中,12.95处单峰归属为A环5位酚羟基,7.88-7.90 与7.54-7.59 处多重峰归属为 B 环的5个氢,6.98处单峰归属为C环3位上的氢,6.75处单峰归属为A环8位上的氢.
同时,区别于黄芩素1H-NMR谱图,化合物(4)1H-NMR谱图中,在2.36-2.38处出现一组新的单峰,归属为主环6与7位乙酰基上的6个氢.因此,可以鉴定成功合成了化合物(4).
2.2.2 黄芩素衍生物5的表征
对照黄芩素1H-NMR谱图,在化合物(4)的1H-NMR中,12.78处单峰归属为A环5位酚羟基,7.49-7.54 与7.86-7.88 处多重峰归属为 B 环的5个氢,6.63处单峰归属为C环3位上的氢,6.54处单峰归属为A环8位上的氢.
同时,区别于黄芩素1H-NMR谱图,化合物(4)1H-NMR谱图中,高场出现了新的信号,4.24与4.74两处三重峰归属为8元饱和环上分别与氧相连的2个亚甲基的4个氢,1.83与2.06两处多重峰分别归属为8元饱和环上除与氧相连的2个亚甲基的4个氢.因此,可以鉴定成功合成了化合物(5).
3 结 论
(1)通过Mannich反应在黄芩素8位α氢引入了胺甲基类基团,成功合成并鉴定了3个Mannich碱衍生物;该类反应条件温和,制备方法简便,产品收率高,可作为一种向黄芩素母核引入含氮基团,提高黄芩素类化合物水溶性的有效方法.
(2)在伯胺的Mannich反应中要注意伯胺与甲醛的摩尔比,比例变化会得到不同产物.在实验过程中,随着苄胺与甲醛的毫摩尔比由1 1改变为1 2,反应产物的Rf值发生了显著的变化,不再与Mannich碱一致,经1H-NMR鉴定为含六元环的二氢苯并噁嗪衍生物.可能是反应产物Mannich碱进一步与甲醛反应生成N-取代羟甲基化中间体,再脱水环合,得到含六元环的二氢苯并噁嗪衍生物.
(3)针对6、7位羟基进行修饰,利用乙酰基选择性地保护6,7位羟基可以成功合成预期化合物,实验结果表现出较强的选择性.但利用其7-位羟基选择性地与溴取代物反应时效果不佳.
[1] Gao D,Sakurai K,Katoh M.Inhibition microsomal lipid peroxidation by baicalein:A possible formation of an iron baicalein complex[J].Biochem Mol Biol Int,1996,39(2):215-216.
[2] 赵晶.两种抗人免疫缺陷病毒天然产物结构修饰的研究[D].北京:中国协和医科大学,1998.
[3] Hong T,Jin GB,Cho S.Evolution of the antiinflammatory effect of baicalein on dextran sulfate sodium induced colitis in mice[J].Planta Medica.2002,68(3):268-271.
[4] Chen ZY,Su YL,Lau CW.Endothelium-dependent contraction and direct relaxation induced by baicalein in rat mesenteric artery[J].European Journal of Pharmacology,1999,374(1):41.
[5] Inoue T,Jackson E K.Strong anti-prolifentive effects of baicalein in Cultured rat hepatic stellate cells[J].Eur J Pharmcol,1999,378(1):129-135.
[6] Zhang L,Lin G,chang Q.Role of Intestinal First-Pass Metabolism of Baicalein in its Absorption Process[J].Pharmaceutical Research,2005,22(7):1050-1058.
[7] 高燕,顾振纶,蒋小岗,等.黄芩素药理学研究新进展[J].时珍国医国药.2010,21(7):1765-1767
[8] 刘耀武,栗进才,夏成凯.关于天然药物活性成分结构修饰与改造的研究探讨[J].时珍国医国药.2009,20(6):1553-1555.
[9] 李云霞,索全伶.黄芩素的分离纯化与结构表征[J].光谱学与光谱分析.2008,28(8):1895-1899.
猜你喜欢
现代畜牧科技(2021年8期)2021-10-13
今日农业(2020年16期)2020-12-14
基层中医药(2020年7期)2020-09-11
数学物理学报(2020年2期)2020-06-02
世界农药(2019年3期)2019-09-10
中成药(2017年12期)2018-01-19
中学生数理化·高二版(2016年3期)2016-12-26
合成化学(2015年10期)2016-01-17
合作经济与科技(2015年22期)2015-08-09
四川师范大学学报(自然科学版)(2015年3期)2015-02-28
