
微波灰化-液相色谱-电感耦合等离子体质谱联用测定干食用菌中的三价铬和六价铬
2014-10-22 12:57:26倪张林汤富彬屈明华莫润宏
色谱 2014年2期
倪张林,汤富彬,屈明华,莫润宏
(中国林业科学研究院亚热带林业研究所,国家林业局经济林产品质量检验检测中心(杭州),浙江 富阳 311400)
铬(Cr)在自然界中主要以三价铬(Cr(Ⅲ))和 六价铬(Cr(Ⅵ))这两种形态存在。Cr(Ⅲ)可激活胰岛素,参与机体糖、脂肪、蛋白质的代谢,能增强胆固醇的分解和排泄,防止动脉硬化,促进氨基酸的转运和蛋白质的合成,促进人体的生长发育,但较高剂量的Cr(Ⅲ)仍表现出细胞毒性反应,长期的累积毒性还有待于进一步的研究[1];Cr(Ⅵ)可以以和Cr2的形态通过带负电荷的细胞膜,并且促使氧化,从而导致病变发生,具有致癌和诱发基因突变的作用。
植物体对铬的富集能力较弱,我们日常食用的蔬菜水果,其总铬含量一般在0.05 mg/kg以下[2],即使是种植在铬污染地区的植物铬含量也处在较低水平[3]。分析近几年的文献报道[4,5],我们发现:食用菌中铬含量处在较高水平[5],但是针对食用菌中铬形态的研究却未见报道。因此,为了对食用菌中铬的营养与安全做出正确的评价,分别测定其中的Cr(Ⅲ)和Cr(Ⅵ)十分有必要。
目前国内外测定铬形态的分析方法主要有原子吸收光谱法[6-8]、分光光度 法[9,10]、离子色谱法(IC)[11]、离子色谱与电感耦合等离子体质谱联用法(IC-ICP-MS)[12]和高效液相色谱与电感耦合等离子体质谱联用法(HPLC-ICP-MS)[13-17]等。这些方法在测定水质、尿样和牛奶等液体样品中已有一些应用,尤其是HPLC-ICP-MS方法,其采用乙二胺四乙酸(EDTA)与Cr(Ⅲ)配合形成稳定的Cr(Ⅲ)-EDTA,使Cr(Ⅲ)和Cr(Ⅵ)能够在不同类型的色谱柱上实现分离,并且可保证Cr(Ⅲ)在碱性环境下的稳定性[13];但是针对生物质固体样品,采用传统的前处理方法如浸提、搅拌和超声提取等方法进行样品前处理,提取效率低,同时在提取过程中,铬形态的稳定性得不到保证。本实验以日常食用量较大的干食用菌为例,采用微波灰化技术,通过高温消除食用菌样品中的有机物,在这个过程中同时能保证样品中原有的铬形态不发生变化,灰化后的样品用EDTA溶液配合形成Cr(Ⅲ)-EDTA稳定其中的 Cr(Ⅲ),通过优化后的HPLC-ICP-MS方法测定其中的Cr(Ⅲ)和 Cr(Ⅵ)。
1 实验部分
1.1 仪器与试剂
Flexar Binary液相泵(美国PerkinElmer公司);NexIon 300D ICP-MS(美国 PerkinElmer公司);PHOENIX微波灰化系统(美国CEM公司);Milli-Q超纯水系统(美国MILLIPORE公司)。50 mL石英坩埚以及其他器皿用酸(30%硝酸)浸泡24 h后,用超纯水冲洗晾干备用。
硝酸,优级纯,德国Merck公司;氨水,优级纯,德国CNW公司;EDTA二钠盐,分析纯,广东西陇化工有限公司;Cr(Ⅲ)和Cr(Ⅵ)形态标准溶液,1000 mg/L,美国 Sigma-Aldrich公司;质谱调谐液,PerkinElmer公司。
1.2 样品前处理
干食用菌样品采用旋风磨粉碎,过40目筛,称取过筛样品0.2 g(精确至0.0001 g)于石英烧杯中,放入微波灰化炉中,设定程序为:10 min升至300℃,保持30 min,10 min升至600℃,保持2 h;运行完毕冷却后取出样品,用10 mL 1%硝酸少量多次将样品洗入50 mL离心管中,再用0.5 mmol/L EDTA二钠溶液反复洗涤至离心管中,用氨水(1∶1,v/v)调节pH值为6,定容至50 mL;将定容好的样品放置在60℃的水浴中静置60 min,使EDTA与Cr(Ⅲ)充分配合,取出冷却后上机备用,同时做样品空白和加标回收试验。
1.3 标准溶液的配制
用0.5 mmol/L EDTA二钠(pH 6)稀释Cr(Ⅲ)和Cr(Ⅵ)标准溶液至1 mg/L,然后置于水浴中于60℃下保持60 min,取出冷却后作为标准储备液。标准使用液现配现用。
1.4 仪器条件
1.4.1 色谱条件
Hamilton PRP-X100阴离子交换柱(250 mm×4.6 mm,10 μm),流动相为 60 mmol/L 硝酸(pH 9.3,氨水调节),流速为1.0 mL/min,进样量为20 μL。
1.4.2 ICP-MS 条件
射频(RF)功率:1500 W;等离子气(Ar)流速:13 L/min;反应气(O2)流速:0.4 mL/min;Rpq(Rejection parameter q)值:0.55;雾化气流速:1.12 L/min。
2 结果与讨论
2.1 灰化条件的选择
传统的马弗炉灰化方法是在样品进入马弗炉之前需要对其进行炭化,操作繁琐并且增加样品被污染的可能性。微波灰化可省去炭化过程,并且炉温冷却迅速,节省了样品前处理时间,因此本实验采用微波灰化对食用菌样品进行前处理;另外,较大的称样量会延长灰化时间,同时也会增加样品溶液中盐分含量,对色谱柱产生不利;综合考虑,选择称样量为0.2 g进行灰化实验。
Cr属于高温元素,为了在较短的灰化时间内得到较好的灰化效果,考察了400℃至900℃下的灰化效果。结果发现,在低于600℃条件下不能将样品较好地灰化,灰分中有较多的黑色炭颗粒;当温度在600~900℃时,灰化后的样品都呈白色,灰化效果较好。为了缩短灰化炉的冷却时间,选择灰化温度为600℃。
为了验证Cr(Ⅲ)和Cr(Ⅵ)在灰化过程中是否会发生相互转化,于空白样品中分别加入10 μg/L的Cr(Ⅲ)和Cr(Ⅵ),通过灰化前处理后上机测试其加标回收率,结果表明:单独进行Cr(Ⅲ)加标回收试验时,Cr(Ⅵ)未检出;单独进行Cr(Ⅵ)加标回收试验时,Cr(Ⅲ)也未检出;Cr(Ⅲ)和Cr(Ⅵ)的加标回收率均达到了85%以上,这说明在灰化过程中Cr(Ⅲ)和Cr(Ⅵ)并未出现相互转化,这与文献报道的结论[8,18]相符合。
2.2 Cr(Ⅲ)-EDTA配合温度的选择
用0.5 mmol/L EDTA二钠溶液(pH 6)配制10 μg/L的Cr(Ⅲ)和Cr(Ⅵ)标准溶液,分别置于40、50、60、70、80 和90 ℃的水浴中 90 min,考察不同温度下标准溶液中Cr(Ⅲ)和Cr(Ⅵ)的稳定性。结果表明,随着温度的升高,Cr(Ⅵ)的响应信号无明显变化,而Cr(Ⅲ)在大于80℃时响应信号下降明显(见图1)。综合考虑选择60℃作为Cr(Ⅲ)-EDTA的配合温度。
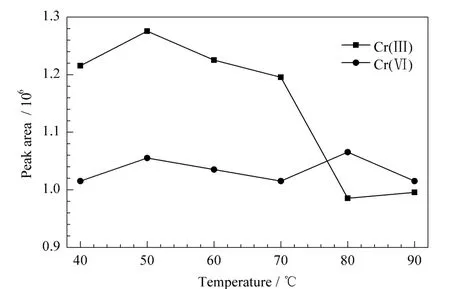
图1 配合温度对Cr(Ⅲ)和Cr(Ⅵ)稳定性的影响Fig.1 Influences of stabilities of Cr(Ⅲ)and Cr(Ⅵ)at different complexation temperatures
2.3 Cr(Ⅲ)-EDTA配合时间的选择
用0.5 mmol/L EDTA二钠溶液(pH 6)配制10 μg/L的Cr(Ⅲ)和Cr(Ⅵ)标准溶液,分别置于60℃水浴中10、20、30、60、90 和 120 min,考察不同温度下标准溶液中Cr(Ⅲ)和Cr(Ⅵ)的稳定性。结果表明,随着配合时间的延长,整个过程中Cr(Ⅵ)响应信号无明显变化,而Cr(Ⅲ)在大于60 min时响应信号才趋于稳定(见图2),表明此时Cr(Ⅲ)与EDTA已完全配合,因此选择60 min作为Cr(Ⅲ)-EDTA的配合时间。
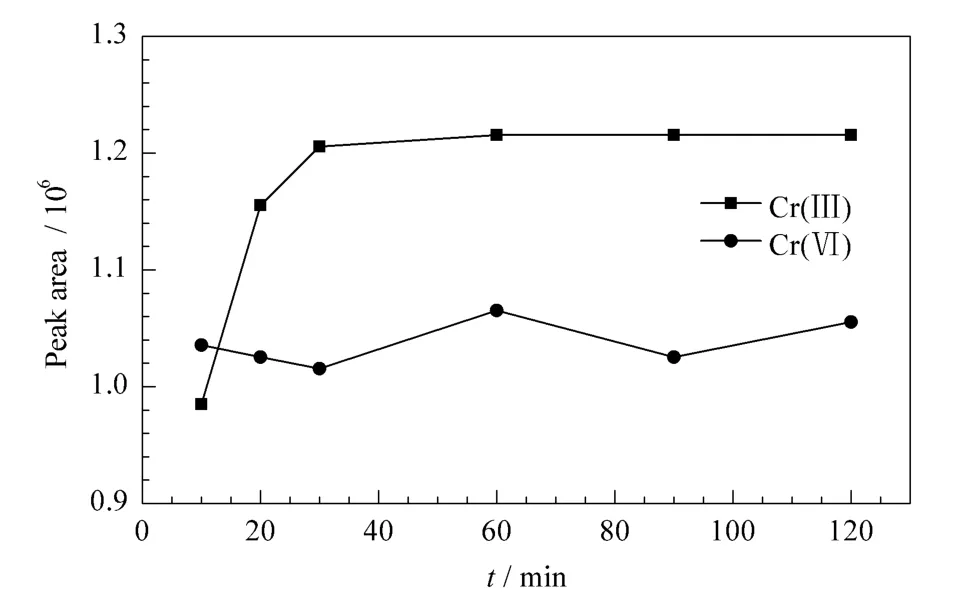
图2 配合时间对Cr(Ⅲ)和Cr(Ⅵ)稳定性的影响Fig.2 Influences of stabilities of Cr(Ⅲ)and Cr(Ⅵ)at different complexation times
2.4 仪器条件的选择
2.4.1 色谱柱及流动相的选择
文献[13-17]报道C18、C8硅胶柱和阴离子色谱柱均可实现Cr(Ⅲ)和Cr(Ⅵ)的分离,但使用C18、C8硅胶柱分离时流动相中需要加入离子对试剂,对色谱柱损伤较大;而阴离子色谱柱应用广泛,耐受性强,因此本实验采用阴离子色谱柱分离Cr(Ⅲ)和Cr(Ⅵ)。
流动相采用60 mmol/L硝酸(pH 9.3)。加大流动相浓度可加快这两种物质的出峰时间,当流动相浓度大于60 mmol/L时导致Cr(Ⅲ)-EDTA和Cr(Ⅵ)的保留时间重叠;另外,过高的pH值也会导致保留时间重叠,过低的pH值易导致Cr(Ⅲ)-EDTA出峰时间延长。
2.4.2 质谱干扰的消除
铬元素在自然条件下有6种同位素,丰度最大的为52Cr(丰度83.8%),但52Cr受质谱干扰严重。实验中发现:采用标准模式(STD)测试,52Cr信号完全被噪声信号湮没,为了得到理想的灵敏度,实验加入NH3作为反应气,在动态反应(DRC)模式下使NH3与干扰物反应形成其他质量数的物质,从而避免质谱干扰。通过仪器优化后,当NH3的流量为0.4 mL/min、Rpq值为0.55时,52Cr的响应值最大,同时背景干扰最低。图3为加入NH3后,检测52Cr得到的Cr(Ⅲ)和Cr(Ⅵ)的色谱图。
2.5 线性关系
取适量的标准储备液用流动相配制不同浓度的Cr(Ⅲ)和Cr(Ⅵ)混合标准溶液,在优化的条件下进行检测,确定Cr(Ⅲ)和Cr(Ⅵ)的线性范围、回归方程以及相关系数(r2);按信噪比(S/N)为3及S/N=10分别计算其检出限(LOD)和定量限(LOQ)(见表1)。结果显示:在0.5~100 μg/L范围内,目标物的峰面积(Y)与其质量浓度(X,μg/L)的线性关系良好,r2均达到0.9999以上。
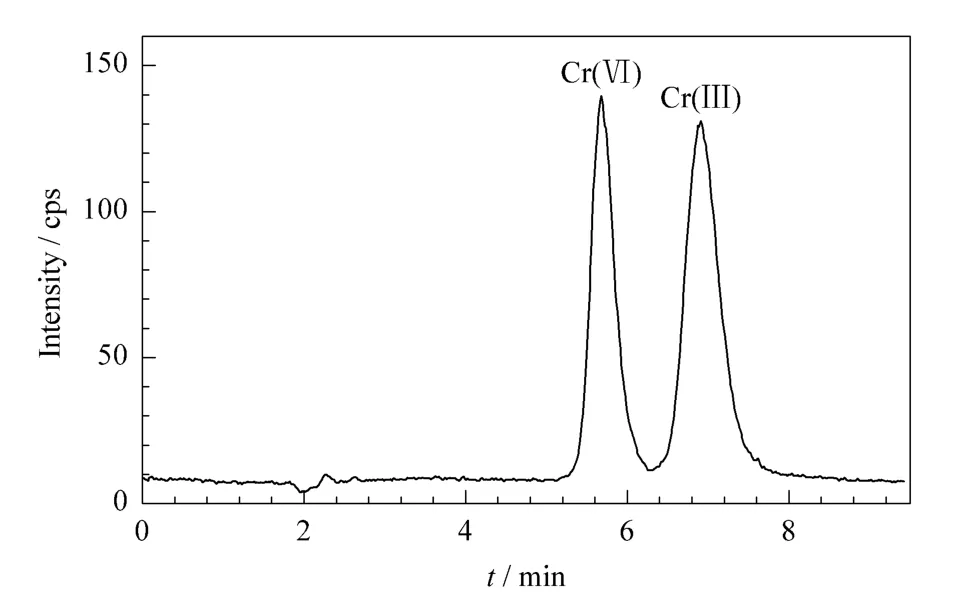
图3 Cr(Ⅲ)和Cr(Ⅵ)标准溶液(10 μg/L)的色谱图Fig.3 Chromatogram of Cr(Ⅲ)and Cr(Ⅵ)standard solution(10 μg/L)

表1 Cr(Ⅲ)和Cr(Ⅵ)的线性方程、相关系数(r2)、线性范围、检出限与定量限Table 1 Regression equations,correlation coefficients(r2),detection limits(LODs)and quantification limits(LOQs)of Cr(Ⅲ)and Cr(Ⅵ)
2.6 方法的回收率与精密度
选用干香菇(mushroom)和干木耳(agaric)做加标回收试验。分别在样品中添加Cr(Ⅲ)和Cr(Ⅵ)混合标准溶液,添加水平分别为1倍LOQ(0.5 μg/L)、10 倍 LOQ(5 μg/L)和 50 倍 LOQ(25 μg/L),按优化后的方法对样品进行前处理和测定,计算平均回收率,结果见表2。由表2可知:Cr(Ⅲ)和Cr(Ⅵ)的平均回收率范围分别为78.0%~90.7%和78.3%~86.4%,RSD分别为1.5%~3.8%和1.9%~3.5%,可满足干食用菌中Cr(Ⅲ)和Cr(Ⅵ)的检测需求和安全评价。
2.7 实际样品测定
随机从市场上抽取6份干食用菌样品进行测试,其中5份样品中Cr(Ⅲ)含量在0.083~0.38 mg/kg之间,均未检出Cr(Ⅵ);1份野生牛肝菌(bolete)样品中Cr(Ⅲ)含量为2.13 mg/kg,Cr(Ⅵ)含量为0.23 mg/kg,其色谱图见图4。
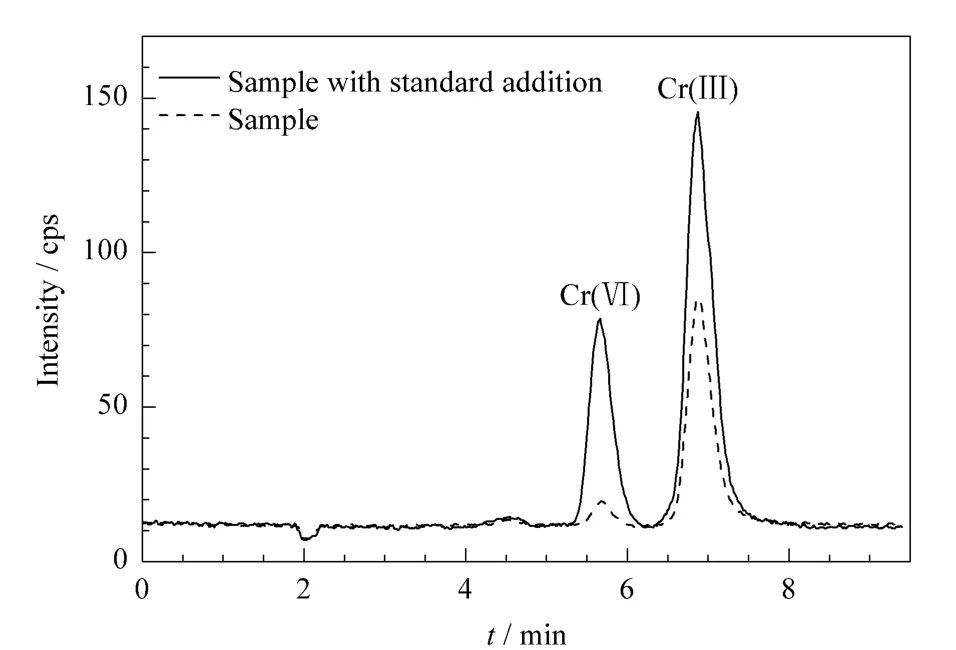
图4 牛肝菌样品及其添加5 μg/L Cr(Ⅲ)和Cr(Ⅵ)标准溶液的色谱图Fig.4 Chromatograms of a bolete sample and the bolete sample spiked with Cr(Ⅲ)and Cr(Ⅵ)solution at 5 μg/L
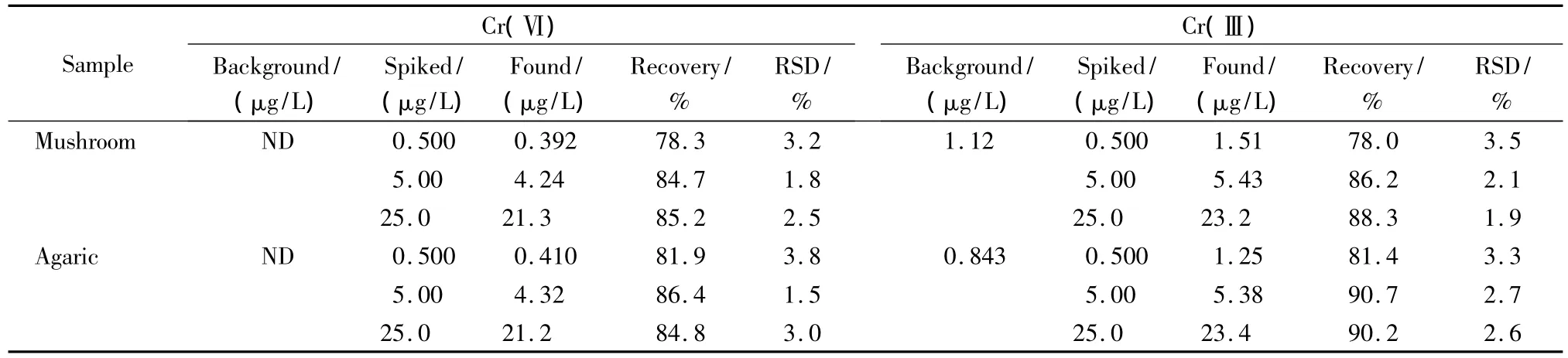
表2 干食用菌中Cr(Ⅲ)和Cr(Ⅵ)的加标回收率与相对标准偏差(n=6)Table 2 Recoveries and RSDs of Cr(Ⅲ)and Cr(Ⅵ)spiked in dried edible fungi samples(n=6)
3 结论
本文建立了微波灰化-液相色谱-电感耦合等离子体质谱(LC-ICP-MS)检测干食用菌中三价铬和六价铬的方法。考察了Cr(Ⅲ)-EDTA配合温度和配合时间对Cr(Ⅲ)和Cr(Ⅵ)测定的影响,结果表明,当配合温度在60℃、配合时间为60 min时,Cr(Ⅲ)和Cr(Ⅵ)的响应信号稳定。通过优化色谱条件和质谱条件,能够实现Cr(Ⅲ)和Cr(Ⅵ)的较好分离,同时消除其他近质量数的质谱干扰。该方法稳定、可靠、灵敏,可满足干食用菌中Cr(Ⅲ)和Cr(Ⅵ)的测定。
[1]Owlad M,Aroua M K,Daud W A W,et al.Water Air Soil Poll,2009,200:59
[2]Chen G Y.Journal of Instrumental Analysis(陈国友.分析测试学报),2007,26(5):742
[3]Yang D Q,Zhou Y,Lei S R,et al.Sichuan Environment(杨定清,周娅,雷绍荣,等.四川环境),2008,27(2):71
[4]Kalac P,Svoboda L.Food Chemistry,2000,69(3):273
[5]Zhu F,Qu L,Fan W,et al.Environ Monit Assess,2011,179(1-4):191
[6]Cheng Y A,Gao S B,Wang F,et al.Journal of Northwest Agriculture&Forestry University:Natural Science Edition(程永安,高双斌,王枫,等.西北农林科技大学学报:自然科学版),2004,32(Suppl):91
[7]Lu J S,Xu J J,Tian J Y,et al.Chinese Journal of Applied Chemistry(卢菊生,徐佳佳,田久英,等.应用化学),2010,27(10):1230
[8]Xiao Y B,Wu Y H,Zhang M,et al.Journal of Instrumental A-nalysis(肖亚兵,吴延晖,张曼,等.分析测试学报),2007,26(2):235
[9]Wang J T,Ma W X,Lü S T,et al.Environmental Monitoring in China(汪军涛,马卫兴,吕松涛,等.中国环境监测),1997,13(4):30
[10]Zhang A Y,Zhu Q R.Chinese Journal of Spectroscopy Laboratory(张爱英,朱庆仁.光谱实验室),2013,30(1):38
[11]Yu R P,Hu Z Y,Ye M L,et al.Chinese Journal of Chromatography(虞锐鹏,胡忠阳,叶明立,等.色谱),2012,30(4):409
[12]Zhu M,Lin S M,Yao Q,et al.Journal of Zhejiang University:Science Edition(朱敏,林少美,姚琪,等.浙江大学学报:理学版),2007,34(3):326
[13]Pantsar-Kallio M,Manninen P K.Fresenius J Anal Chem,1996,355(5/6):716
[14]Namiesnik J,Rabajczyk A.Crit Rev Env Sci Tec,2012,42(4):327
[15]Burbridge D J,Koch I,Zhang J,et al.Chemosphere,2012,89(7):838
[16]Wang H J,Li Y H,Feng W Y,et al.Chinese Journal of Analytical Chemistry(王华建,黎艳红,丰伟悦,等.分析化学),2009,37(3):433
[17]Andrle C M,Jakubowski N,Broekaert J A C.Spectrochim Acta Part B,1997,52(2):189
[18]Kovács R,Béni Á,Karosi R,et al.Food Chem,2007,105(3):1209
猜你喜欢
今日农业(2020年23期)2020-12-31 09:00:42
今日农业(2020年22期)2020-12-25 02:30:40
化工设计通讯(2020年10期)2020-09-17 14:43:16
今日农业(2020年24期)2020-03-17 08:58:04
中国资源综合利用(2017年2期)2018-01-22 02:45:01
Cancer Biology & Medicine(2016年4期)2017-01-13 01:54:45
中国氯碱(2016年9期)2016-11-16 03:07:39
分析测试学报(2015年7期)2016-01-13 06:19:23
江西理工大学学报(2015年3期)2015-12-22 05:26:17
湖南农业科学(2014年5期)2014-02-27 14:29:20
