
慢性进行性眼外肌瘫痪1例报告并文献学习
2014-09-26 02:17:02肖君陈博田代实
神经损伤与功能重建 2014年1期
肖君,陈博,田代实
慢性进行性眼外肌瘫痪1例报告并文献学习
肖君,陈博,田代实
慢性进行性眼外肌瘫痪;破碎红纤维;晶格状包涵体;线粒体DNA
tiands@tjh.tjmu.edu.cn
慢性进行性眼外肌瘫痪(chronic progressive extemalophthalmoplegia,CPEO)是一种以慢性进行性眼睑下垂及眼球活动障碍,特别是上转障碍为特征的线粒体肌病[1]。部分患者可有咽部肌肉和四肢无力,而且对新斯的明试验不敏感。本文报告1例如下。
1 病历资料
增多,线粒体大小不一,形态不规则,线粒体嵴结构模糊或呈同心圆状排列,基质密度增高,晶格状包涵体(mitochondrialparacrystaline inclusion bodies,MIBs)多见,考虑线粒体肌病,见图3。患者基因检测未见多聚腺核苷酸结合蛋白 2(polyadenylate-binding protein 2,PABP2)的 GCG 重复突变,见图4,未行单基因突变检测。
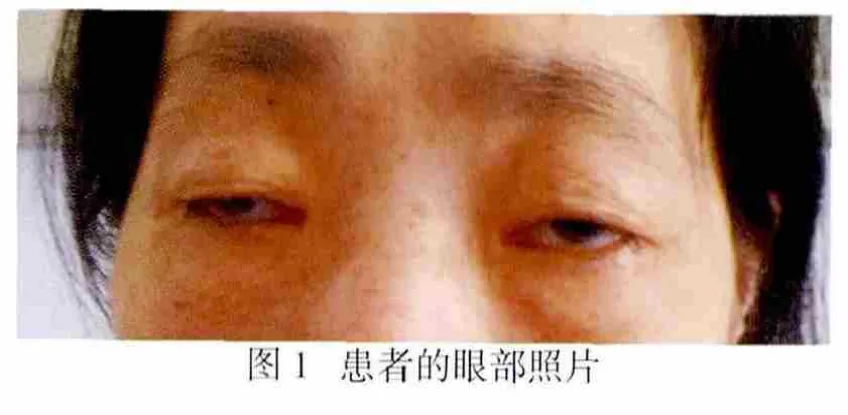
患者,女,43岁,因“眼睑下垂20余年,进行性四肢乏力十余年,吐词不清4、5年”于2012年6月1日入院。患者20年前无明显诱因出现左眼睑下垂,行提上睑术。后逐渐出现双眼睑下垂、进行性消瘦、四肢乏力,近4年症状进行性加重,并出现吞咽困难、构音障碍、饮水呛咳,见图1。上述症状无波动及疲劳后加重现象,日常生活能自理。多次于我院门诊就诊,行新斯的明试验阴性,肌电图示左侧三角肌偶见失神经电位,运动单位电位小,提示肌源性疾病的可能性较大;重复频率电刺激(repetitive nerve stimulation,RNS)示M波幅无显著改变。既往史、家族史均无特殊。查体:内科检查无异常。神经科专科体检:神清,查体配合,消瘦,双侧瞳孔等大等圆,直径3mm,对光反射存在,双眼睑下垂,双眼球固定位,各方向活动受限;双侧额纹对称,双侧眼轮匝肌闭合力弱,鼻唇沟对称,鼓腮、露齿力弱,伸舌居中,双侧软腭运动减弱,咽反射减弱;颈软,克、布氏征(-),四肢肌力5-级,肌张力正常,躯干肌及四肢近端肌萎缩明显;Gower征(+),病理征(-),感觉检查(-),指鼻试验(-)。实验室检查:血尿常规、血生化、甲状腺功能、凝血功能均无明显异常,血乳酸 3.6mmol/L(正常值为 0.5~2.2mmol/L)。胸片未见明显异常。心电图正常范围。颅脑MRI平扫未见明显异常。眼底镜检查未见视网膜色素变性。肌活检HE染色见肌纤维大小形态各异,有较多破碎红纤维(ragged-red fiber,RRF)样肌纤维;Gomori染色示部分肌纤维可见异常空泡,有较多RRF样肌纤维;SDH染色见约30%肌纤维呈RRFs样外观;COX染色示约30%肌纤维异常浅染,见图2。电镜示大多数肌细胞肌结构正常,部分肌细胞可见局灶性肌丝溶解,部分细胞肌膜下或肌溶灶间线粒体
2 讨论
CPEO是最常见的线粒体肌病之一,任何年龄均可发病,儿童或青少年,即30岁以前起病多见,多为散发,亦有家族性报道。主要表现为慢性、双侧性、进行性的上睑下垂和眼球运动障碍,可伴咽部及肢体肌无力。由于双侧眼肌均受累,且眼肌麻痹极缓慢,故较少出现复视[2]。中枢神经系统功能障碍如痴呆、痫样发作或卒中样发作亦少见[3,4]。
根据Walker等[5]在1996年发表的线粒体脑肌病的主要诊断指标,符合线粒体脑肌病各综合征的临床表现,肌活检RRF>2%,50岁以下肌活检COX(-)肌纤维数量>2%,发现相关的nDNA或m tDNA异常可确诊本病。本例为女性患者,43岁,儿童期起病,表现为进行性双眼睑下垂直至眼球固定,无晨轻暮重,并出现进行性咽部及四肢肌肉无力,新斯的明及疲劳试验(-),符合CPEO临床表现;肌肉活检发现较多RRF,COX染色发现30%COX(-)肌纤维,故CPEO诊断明确。
肌肉活检发现RRF和COX阴性纤维是线粒体肌病最具特征性的病理改变,但没有这些病理发现并不能排除诊断[6,7]。该患者肌肉活检Gomori染色及SDH染色发现较多RRF纤维,且HE染色可见散在单个异常深染纤维,与Gomori染色所显示的RRF一致,COX染色见30%肌纤维异常浅染,呈马赛克样表现。RRF是线粒体发生结构功能异常增生聚集所致,因此除Gomori染色外,反映线粒体增生的SDH染色同样也能发现RRF,且比Gomori染色更为敏感[8]。COX是一个重要的呼吸链酶,CPEO可有呼吸链酶受抑制的表现,呼吸链酶复合体Ⅳ(COX)的缺陷导致COX(-)肌纤维的产生[9]。
虽然CPEO的诊断依赖于光镜发现RRF,但可因免疫组化染色失败或线粒体突变程度不同而缺乏RRF,电镜则弥补了光镜的不足。电镜能清楚地观察到线粒体超微结构,特别是M IBs有助于对本病的诊断[10]。该患者电镜示线粒体堆积,形态异常,M IBs多见。M IBs的主要成分是线粒体肌酸激酶(Mitochondrial creatine kinase,M i-CK),由于CPEO患者线粒体ATP生成障碍,M i-CK过度表达,线粒体内堆积而形成,是不可逆性病变的标志[11]。
该患者空腹血乳酸水平增高,血乳酸在线粒体疾病中可增高,这对作出CPEO诊断及进一步行肌肉活检和基因检测有提示作用。但有报道表明仅40%线粒体疾病患者空腹血乳酸升高[12],因此乳酸不能作为筛查指标。
CPEO的发病机制尚不清楚,线粒体DNA(m tDNA)突变逐渐被认为是其主要的病因。m tDNA是一条长度为16 569碱基对的双链分子,共编码氧化磷酸化过程中的13个蛋白亚基、2个核糖体RNA和所有转运RNA[13]。m tDNA的缺失导致线粒体功能的缺陷,尤其是在肌肉、脑组织和心脏中,细胞内突变m tDNA越多,线粒体功能缺失越多[14]。眼外肌由于线粒体的数目比其他骨骼肌的数目高出很多,因此更易受影响,这也可以解释为何CPEO最易累及眼外肌。m tDNA突变类型可为点突变、基因重排(重复、单基因缺失或多基因缺失)及m tDNA丢失[14]。CPEO常为散发性,不遗传给后代,亦有三分之一为家族遗传性,其遗传方式可为母系遗传或常染色体遗传[15]。散发的CPEO多由m tDNA的单基因缺失引起,这种突变常随机发生在卵子受精后,因而不传给后代[16,17]。m tDNA点突变常引起母系遗传的CPEO,最常见的突变位点是A3243G[18,19]。常染色体遗传的CPEO多归因于m tDNA的多基因缺失,核DNA的原发缺陷引发m tDNA复制过程中的继发多基因缺失[20]。
CPEO临床表现多种多样,需与多种神经系统疾病如眼咽型肌营养不良(oculopharyngealmuscular dystrophy,OPMD)、Kearns-Sayre综合征(Kearns-Sayre syndrome,KSS)、眼肌型重症肌无力相鉴别。OPMD多在20~30岁缓慢起病,最初表现为双眼睑下垂,其后累及眼外肌,肌肉病理可见少量RRF,易与CPEO混淆。但OPMD为多聚腺核苷酸结合蛋白2(PABP2)的GCG重复突变增加所致[21],而CPEO多为m tDNA的单一大片段缺失或点突变所致,因此突变基因分析可有助于两者鉴别。KSS是线粒体疾病的另一种类型,一般在20岁之前发病,具有CPEO表现,同时还应有视网膜色素变性及心脏传导阻滞。其他神经系统异常包括小脑性共济失调、脑脊液蛋白增高、神经性耳聋和智能减退等。且该病进展快,多在20岁前死于心脏病。本例患者已43岁,缓慢起病,眼底镜检查、共济运动检查正常,无明显心脏受累的表现,可鉴别。CPEO容易和眼肌型重症肌无力相混淆,MG患者有晨轻暮重表现,新斯的明试验及疲劳试验阳性,肌电图重复频率电刺激为高、低频重复电刺激波幅均降低,本例患者多次新斯的明试验阴性,服用胆碱酯酶抑制剂无效,肌肉活检见RRF,均可鉴别。
在治疗上,迄今为止尚未发现具有治愈性疗效的药物及方法,目前主要是对症治疗,饮食、药物及物理治疗相结合,辅酶Q10及B族维生素可使血乳酸水平降低,但该治疗方法疗效尚未肯定。

图2 患者肌活检染色(×400)
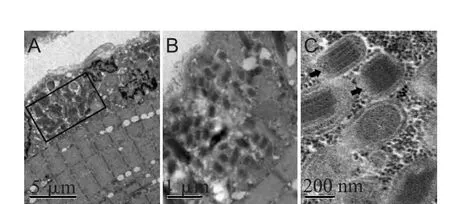
图3 患者肌活检电镜照片

图4 患者PABPN1基因GCG重复检测
综上所述,CPEO是线粒体肌病的一种常见类型,以进行性上睑下垂及眼球活动障碍为临床特点,m tDNA突变可能是其主要病因,迄今为止尚无特效治疗。对临床上以眼外肌瘫痪为主要表现,新明试验阴性、肌电图RNS无改变的患者,应考虑到该病的可能性,行肌肉活检检查,如有条件可行基因检测,力求对CPEO患者进行早期诊断,防止误诊、漏诊的发生。
[1]Hart PE,De Vivo DC,Schapira AH.Clinical features of the mitochondrial encephalomyopathies//Shapira AH,DiMauro S,eds.MitochondrialDisorders in Neurology 2. Woburn: Butterworth-Heineman,2002:35-68.
[2]Wallace DK,Sprunger DT,Helveston EM,et al.Surgicalmanagement of strabismusassociated with chronic progressive externalophthalmop legia[J].Ophthalmology,1997,104:695-700.
[3]Hammans SR,Sweeney MG,Brockington M, et al. Mitochondrial encephalopathies:moleculargenetic diagnosis from blood samples[J].Lancet,1991,337:1311-1313.
[4]Truong DD,Harding AE,Scavallini F,etal.Movement disorders inMitochondrial myopathies:a study of nine caseswith two autopsy studies[J].Mov Disord,1990,5:109-117.
[5]Walker UA,Collins S,Byrne E.Respiratory chain encephalomyopathies:a diagnostic classification[J].Eur Neurol,1996,36:260-267.
[6]Schaefer AM,Blakely EL,GriffithsPG,etal.Ophthalmoplegia due toMitochondrial DNA disease:the need forgenetic diagnosis[J].Muscle Nerve,2005,32:104-107.
[7]Pineda M,Playan-Ariso A,Alcaine-Villarroya MJ,etal.Fam iliar chronic progressive externalophthalmoplegia ofMitochondrialorigin[J].Rev Neurol,2004,38:1023-1027.
[8]Hammans SR,Sweeney MG,Holt IJ,et al.Evidence for intramitochondrial complementation between deleted and normalmitochondrial DNA in some patientswithMitochondrial myopathy [J].JNeurol Sci,1992,107:87-92.
[9]Bau V,Zierz S.Update on chronic progressive external ophthalmoplegia[J].Strabismus,2005,13:133-142.
[10]李天炼.线粒体肌病的病理分析[J].海南医学院学报,2010,16:1161-1163.
[11]OcGorman E,Piendl T,Muller M,et al.Mitochondrial intermembrane inclusion bodies:The common denominator between human Mitochondrialmyopathies and creatine depletion,due to impairmentof cellular energetics[J].Mol Cell Biochem,1997,174:283-289.
[12]Jackson MJ,Schaefer JA,Johnson MA,etal.Presentation and clinical investigation of Mitochondrial respiratory chain disease.A study of 51 patients[J].Brain,1995,118:339-357.
[13]Anderson S,Bankier AT,Barrell BG,etal.Sequence and organization of the human Mitochondrial genome [J].Nature,1981,290:457-465.
[14]Ryan L.Davis,Carolyn M.Sue.The genetics ofMitochondrial disease[J].Sem in Neurol,2011,31:519-530.
[15]Caballero PE,Candela MS,A lvarez CI,etal.Chronic progressive external ophthalmoplegia:A report of 6 cases and a review of the literature[J]. Neurologist,2007,13:33-36.
[16]Holt IJ,Harding AE,Cooper JM,etal.Mitochondrialmyopathies:clinicaland biochemical features of 30 patientswithmajor deletions ofmuscleMitochondrial DNA[J].Ann Neurol,1989,26:699-708.
[17]LaforetP,Lombes A,Eymard B,etal.Chronic progressive external ophthalmoplegiawith ragged-red fibers:clinical,morphological and genetic investigations in 43 patients [J].Neuromusc Disord,1995,5:399-413.
[18]Moraes CT,Ciacci F,Silvestri G.Atypical clinical presentations associated with the MELASMutation at position 3243 of human Mitochondrial DNA[J].Neuromusc Disord,1993,3:43-50.
[19]Hansrote S,Croul S,Selak M,et al.External ophthalmoplegia with severe progressivemultiorgan involvement associated with the m tDNA A3243G mutation[J].J Neurol Sci,2002,197:63-67.
[20]DeschauerM.Mitochondriale enzephalomyopathien [J].Psychoneurology,2003,29:108-112.
[21]Kim YJ,Noguchi S,Hayashi YK,et al.The productof an oculopharyngealmuscular dystrophy gene,poly(A)-binding protein2,interacts with SKIP and stimulates muscle-specific gene expression[J].Hum MolGenet,2001,10:1129-1139.
R741;R746.9
A
DOI10.3870/sjsscj.2014.01.022
华中科技大学同济医学院附属同济医院神经内科 武汉430030
2012-08 -13
田代实
猜你喜欢
今日农业(2021年5期)2021-11-27 17:22:19
现代畜牧科技(2021年5期)2021-07-20 08:07:40
中国民间疗法(2021年5期)2021-06-09 09:21:22
兽医导刊(2016年6期)2016-05-17 03:50:27
中国医疗美容(2015年2期)2015-07-19 10:11:59
中国医疗美容(2015年2期)2015-07-19 10:11:58
中国医疗美容(2015年1期)2015-07-12 10:06:40
中国当代医药(2015年26期)2015-03-01 02:07:11
实用临床医学(2014年6期)2014-02-28 09:18:32
实用医药杂志(2012年6期)2012-04-21 10:38:42
